Analyse chimique
Méthodes et techniques instrumentales
- Par Francis Rouessac,
- Annick Rouessac,
- Avec la collaboration de Daniel Cruché,
- Claire Duverger-Arfuso
- et Arnaud Martel
Sciences Sup
2019
576 pages
Citer cet ouvrage
- ROUESSAC, Francis,
- ROUESSAC, Annick,
- Avec la collaboration de CRUCHÉ, Daniel,
- DUVERGER-ARFUSO, Claire
- et MARTEL, Arnaud,
- Rouessac, Francis.,
- et al.
- Rouessac, F.,
- Rouessac, A.,
- Avec la collaboration de Cruché, D.,
- Duverger-Arfuso, C.
- et Martel, A.

L’expérience de base en chromatographie.
a) Les ingrédients nécessaires (C, colonne, PS, phase stationnaire, PM, phase mobile et Ech., échantillon) ; b) le dépôt de l’échantillon ; c) le début de l’élution ; d) la récupération des produits après séparation.

Principe de l’analyse par chromatographie.
Le chromatogramme, passage obligé de toute analyse chromatographique, est obtenu à partir des variations en fonction du temps d’un signal électrique envoyé par le détecteur. Il est soit présenté en temps réel soit en différé à partir des valeurs instantanées numérisées et stockées. Les logiciels de chromatographie recalculent ces valeurs pour être mises au format désiré. Chromatogramme illustrant la séparation d’un mélange de 3 constituants principaux. Noter l’ordre d’apparition des pics en correspondance avec la position de chaque constituant dans la colonne.


Caractéristiques d’un pic chromatographique idéal.
Signification des trois paramètres classiques et résumé des caractéristiques d’une courbe de Gauss.

Isothermes de distribution.
1) Situation correspondant à l’invariance de l’isotherme de concentration ; 2) situation pour laquelle la phase stationnaire est saturée — la montée du pic est plus rapide que sa descente ; 3) situation inverse : la phase stationnaire retient trop le constituant, le temps de rétention augmente, la montée du pic est moins rapide que sa descente. Pour chaque colonne, les fabricants indiquent quelle est sa capacité limite (en ng/composé) avant déformation du pic. Illustrations de ces trois situations à partir d’extraits de chromatogrammes réels.

Dispersion d’un soluté dans une colonne.
La courbe de gauche correspond à une image isochrone de la concentration du composé élué à l’instant considéré, et le chromatogramme de droite, à la variation de la concentration en sortie de colonne en fonction du temps. tR et σ sont dans le même rapport que L et σL. L’efficacité N peut donc être calculée à partir du chromatogramme en mesurant σ directement. Sur le graphe ci-dessus on trouverait environ 100 plateaux théoriques.

Facteurs de rétention et de séparation (ou facteur de sélectivité) entre deux composés adjacents.
Chaque composé a un facteur de rétention qui lui est propre. α à lui seul, ne permet pas de savoir si la séparation est réellement possible. Sur cette figure, le facteur de séparation est d’environ 1,3.

Facteur de résolution.
Simulation de pics chromatographiques par juxtaposition plus ou moins rapprochée de 2 courbes gaussiennes identiques. Aspect visuel correspondant aux valeurs de R indiquées sur les diagrammes. À partir de R = 1,5 on considère que les pics sont résolus, la vallée entre les pics étant d’environ 2 %.

Effet de la longueur de la colonne sur la résolution.
Expériences faites en chromatographie en phase gazeuse en modifiant seulement la longueur de la colonne capillaire. On illustre ainsi qu’en doublant la longueur de la colonne la résolution est multipliée par 1,41 (adapté d’un document de la société Waters).

Courbe de Van Deemter en chromatographie gazeuse avec indication des domaines propres à A, B et C.
Il existe également une équation semblable à celle de Van Deemter qui fait intervenir cette fois la température : H = A + B/T + C · T.

Le triangle du compromis entre résolution, vitesse et capacité.
Toute analyse chromatographique est soumise à trois critères contradictoires. Si on privilégie un de ces trois critères, les deux autres sont obligatoirement défavorisés. Quand on recherche une bonne sélectivité, on doit se situer près du sommet résolution de ce triangle. La zone ombrée indique le domaine qui correspond à la chromatographie analytique. Celle-ci tire profit des 5 paramètres : K, N, k, α et R.

Méthode de dosage par étalonnage externe.
La précision de cette méthode est améliorée lorsqu’on utilise plusieurs solutions afin de tracer la droite d’étalonnage. Pour l’analyse de traces, il est parfois conseillé, en chromatographie liquide, de remplacer les aires des pics par leurs hauteurs qui sont moins sensibles aux variations de débit de la phase mobile.





Schéma fonctionnel d’un appareil de CPG et réalisations pratiques.
Chromatographe analytique polyvalent (modèle 7000C de la société Agilent Technologies). L’instrument représenté est muni d’un porte-échantillons (carousel), d’un injecteur, d’un échantillonneur automatique et d’un système de détection par spectrométrie de masse (GC-MS). Les deux autres appareils sont des modèles portables (5-15 kg) pour des analyses faites sur le terrain (composés organiques volatils, matières industrielles toxiques). À gauche le Hapsite ER de Inficon et à droite le Tridion 9 de Torion Technologies.

Efficacité en fonction de la nature et de la vitesse linéaire du gaz vecteur.
Les courbes de Van Deemter reliant HEPT et vitesse linéaire du gaz vecteur pour un même composé. Comparaison des viscosités de ces trois gaz. Noter que la viscosité croît avec la température T.

Microseringue pour CPG et principe d’une boucle d’injection installée dans un processus en continu.
Le modèle choisi pour cette illustration a une pointe conique adaptée pour certains septums ou injecteurs automatiques. Dans ce modèle, le piston rentre dans l’aiguille pour libérer la totalité de l’échantillon et éviter tout volume mort. En bas, exemple de boucle d’injection pour gaz ou liquides (cf. également fig. 3.5).

Injecteur à vaporisation directe utilisé pour colonnes remplies.
Le septum type est un disque en élastomère, mais il en existe des variantes plus sophistiquées, dont le “Microseal” Merlin pouvant servir des milliers de fois (reproduit avec l’autorisation de la société Merlin).

Injecteurs.
En haut : à gauche, chambre d’injection avec diviseur (la sortie 2 règle le split); en bas : aspect typique d’un chromatogramme obtenu en mode splitless. Le pic solvant peut occulter une partie des composés, à moins d’utiliser un détecteur sélectif qui ne “voit” pas le solvant.

Injecteur PTV, à température programmable et injecteur à froid « on column ».
Pour permettre de faire des gradients rapides de température, la chambre d’injection est entourée d’une résistance et d’une circulation de gaz froid.

Colonne capillaire.
Représentation d’une colonne capillaire commerciale de 50 m de longueur enroulée sur son support métallique (Document de la Société Alltech) et détail d’une colonne. À cette échelle, l’épaisseur de phase stationnaire serait à peine visible.

Échelle de polarité des PS en CPG et résumé des 3 types de PS.
L’échelle de polarité va du squalane (polarité 0 par définition) à la polarité 100 pour le TCEP (tricyanoethoxypropane). a) Structure des polysiloxanes (silicones), b) polyéthylèneglycols. Nombreuses compositions de phases de ce type, utilisées en imprégnation ou en greffage. c) ex. d’un liquide ionique du type tetraalkylphosphonium dicationique associé à un imide (ici, le IL60) ; Tmax d’utilisation 300 °C.

Exemple de séparation obtenue avec une phase chirale comportant des cyclodextrines greffées.
Sur une colonne chirale les composés à l’état de racémate se dédoublent (alcools 2 et 4). Ce chromatogramme permettrait également de calculer les indices de rétention des composés séparés.

Phases stationnaires chirales en CPG.
Parmi les 3 trois vecteurs chiraux rencontrés en CPG (1et 2, β-cyclodextrines, 3, éthers-couronne 4, diamides), les cyclodextrines sont de loin les plus utilisées. Elles comportent 3 types de sites : A, hydroxyle axial, B, hydroxyle équatorial et C hydroxyméthyle dont les réactivités sont suffisamment différentes pour permettre des réactions sélectives et obtenir ainsi une cinquantaine de phases (ex. 2, phase cycloSil-B, non greffée de la Soc. Chromoptic). Chromatogrammes partiels d’extraits naturels montrant la séparation des isomères optiques de la carvone.

Analyses de gaz.
À gauche, l’un des tout premiers chromatogrammes, obtenu point par point, représentant un mélange d’air, d’éthylène et d’acétylène séparés sur gel de silice (E.Cremer et F.Prior, Z. Elektrochem. 1951, 55, 66). À droite, une analyse de gaz sur colonne PLOT moderne (reproduit avec l’autorisation de la société Supelco).

Détecteurs FID (a) et NPD (b).
Le gaz d’appoint (ou gaz de remplissage) est utile lorsque le débit de la colonne capillaire est trop petit. Les électromètres permettent de mesurer des intensités qui seraient trop faibles pour un galvanomètre. Nombreuses variantes suivant les constructeurs.

Détecteur à conductibilité thermique.
À gauche agencement montrant la double circulation du gaz vecteur. À droite schéma d’un bloc catharométrique avec le principe de son raccordement électrique dans un montage du type pont de Wheatstone.

Détecteur ECD et PID.
Le PID comporte un filtre dont le choix permet de sélectionner l’énergie des photons afin que seuls les analytes soient les seules espèces ionisées à l’exclusion des molécules du gaz vecteur (ex ; LiF pour 11,8 eV ou saphir pour 8,4 eV). Le mécanisme de l’ionisation est réversible.

Association de trois détecteurs en série.
À la sortie d’une colonne capillaire on peut installer en série ou en parallèle, selon que le détecteur détruit ou non l’échantillon, plusieurs détecteurs de principes différents. Chromatogrammes du mélange injecté, obtenus en sortie de chaque détecteur. On remarquera que la sélectivité varie beaucoup d’un détecteur à un autre.
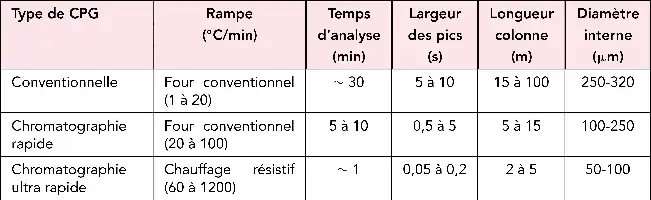

Chromatogramme « ultra-rapide ».
À gauche, séparation de quelques composés aromatiques (chromatographie « rapide » d’après un document de Thermo Electron Corp. ; À droite exemple de chromatogramme obtenu dans des conditions de chromatographie « ultra-rapide » d’après un document de Aviv Analytical.

Droite de Kovats.
Chromatogramme en régime isotherme d’une série de 5 n-alcanes (C10-C14) et droite de Kovats correspondante pour la phase stationnaire et les conditions d’analyse précisées.

Indices de rétention de Kovats (I = 100 nX) sur une colonne en régime isotherme.
Le nombre de carbones équivalent nX, est trouvé à partir du logarithme du temps de rétention réduit \begin{equation}t_{R(X)}^{\prime}\end{equation}. Le chromatogramme correspond à l’injection d’un mélange de 4 n-alcanes et 2 hydrocarbures aromatiques. Les valeurs en italiques correspondent aux temps de rétention en secondes. En injectant périodiquement ce même mélange, la modification des indices de Kovats de ces hydrocarbures permet de suivre l’évolution des performances d’une colonne. En programmation de température on peut encore tracer cette droite en utilisant une formule corrigée, mais la précision est moins bonne.
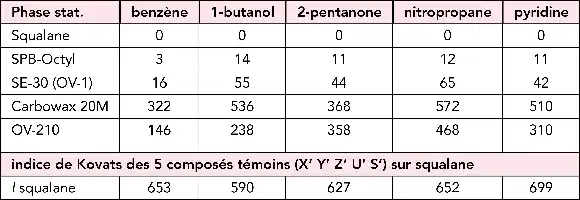

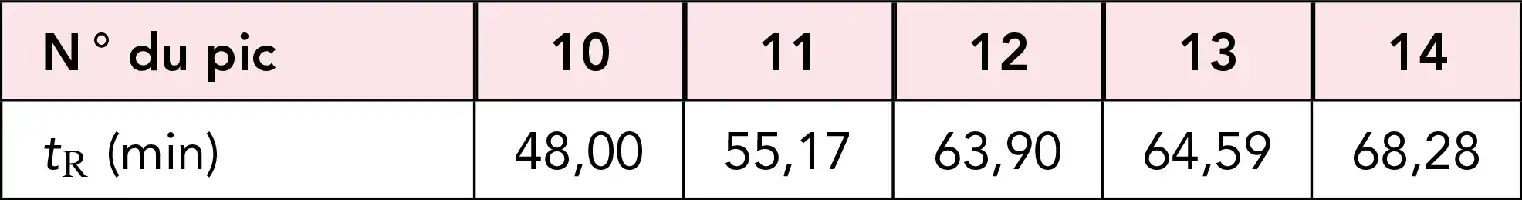

Schéma d’une installation de CLHP. Un exemple de réalisation de type modulaire.
La présentation en colonne des différents modules est commune à de très nombreux modèles concurrents. Ici le chromatographe modèle Agilent 1200 (reproduit avec l’autorisation de la société Agilent Technologies), comporte un injecteur automatique permettant un fonctionnement en continu et une colonne thermostatée pour améliorer la reproductibilité des séparations.

Principe de fonctionnement simplifié d’une pompe à deux têtes en série.
Partant de l’instant où le clapet de sortie b du cylindre A vient de se fermer et le clapet d’entrée a vient de s’ouvrir, le piston de A recule pour remplir la chambre A. Pendant ce temps, c est fermé et d ouvert : le piston de B avance pour chasser la phase mobile vers la colonne. Le volume déplacé par le piston de B est deux fois plus petit que le volume aspiré par le piston de A. Arrivé au fond de sa course, a se ferme, b et c s’ouvrent. Le piston de A chasse le contenu du cylindre A (comme représenté sur la figure) dont la moitié part vers la colonne, l’autre moitié remplissant le cylindre B qui recule. Entre les deux cylindres est placé un amortisseur de pulsations. En bas, variations de débit attendues d’un tel ensemble en fonction des cycles et principe d’un dégazeur.

Exemple de configuration pour gradient haute pression.
Les pompes sont appelées binaires, ternaires, quaternaires suivant le nombre de solvants qu’elles peuvent mélanger (ici quaternaire).

Vanne d’injection pour CLHP et boucles assorties.
Vanne vue de l’arrière (vanne à 6 entrées/sorties avec une boucle raccordée) et assortiment de boucles de différents volumes (reproduit avec l’autorisation de la société Rheodyne Inc.).

Injection avec une boucle.
a) Remplissage de la boucle. Dans cette étape, la seringue est introduite à la position n °4 ; b) injection dans la colonne (noter la nouvelle position de la manette). Vanne modèle 7125. Les vannes sont motorisables (reproduit avec l’autorisation de la société Rheodyne Inc.)

Colonne standard et précolonne de CLHP.
Aspects extérieurs éclatés et assemblage d’une colonne. La phase stationnaire est maintenue entre deux disques poreux. La surface interne du tube est rendue inerte par un traitement de passivation, ou par un chemisage adapté. La précolonne, périodiquement changée, évite le colmatage de la colonne. Sur ce schéma, ont été figurés les 4 types de colonnes de diamètres différents. En admettant que le remplissage de ces 4 colonnes soit de même nature et de même porosité, on voit l’avantage que présente le passage de la CLHP classique (à gauche) à la CLHP-capillaire ou à la nano-CLHP (à droite). Plus le diamètre de la colonne est étroit, plus la consommation de phase mobile diminue, et plus la sensibilité (ici théorique) augmente.

Les gels de silice pour CLHP.
Les phases à noyau dur conduisent à des améliorations notables comme le montrent les 2 courbes de Van Deemter comparant les phases 2 et 3 (extrait d’une notice Agilent concernant les phases 2,7 μm Poroshell-C18 à noyau dur et 1,8 μm Zorbax-C18 totalement poreuse). Les macropores et les mésopores des colonnes monolithiques assurent la circulation de la phase mobile et les micropores les équilibres de concentration.

Nature de la phase stationnaire et efficacité.
La HEPT décroît avec la taille des particules, mais à débit identique, la perte de charge augmente. Les colonnes monolithiques améliorant les transferts de masse entre les phases, apparaissent supérieures. Essais réalisés avec des colonnes de 5 cm remplies d’une phase stationnaire RP-18 de même fabrication et en prenant le naphtalène comme composé test.

Phénomènes d’adsorption et de partage.
L’adsorption est un phénomène d’interface à la différence de l’absorption.

Évolution des performances en chromatographie liquide
Les quatre courbes de Van Deemter reproduites illustrent les progrès technologiques des phases stationnaires au cours des dernières décennies. Les colonnes ont gagné en efficacité tout en permettant des analyses plus rapides avec une moindre consommation de PM (d’après un document de la société Waters concernant les colonnes Acquity).

Formation d’organosilanes greffés à l’interface du gel de silice.
Représentation de quelques groupements monomériques ou polymériques rencontrés en surface du gel de silice. Détail d’une particule chevelue. L’enchaînement Si-O-Si-C est plus stable que Si-O-C. On peut atteindre, pour les revêtements polymériques, plus de 15 % de carbone après greffage sur les silanols accessibles. En bas, un exemple de phase comportant un greffon dipolaire (phase HILIC). D’autres réactions sont aussi utilisées (hydrosylylation en particulier).

Exemples de phases stationnaires chirales utilisées en CLHP.
Séparation des énantiomères de l’oxyde de trans-stilbène (reproduit d’après un document de la société Macherey-Nagel). Sélecteur chiral à base d’amylose ou à base de β-cyclodextrine, substituées sous forme de carbamates.

« Force » des solvants utilisés comme phases mobiles.
On peut, en mélangeant plusieurs solvants, ajuster le pouvoir d’élution de la phase mobile. On notera que la viscosité, donc la pression en tête de colonne, varie selon la composition de la phase mobile.

Polarités de quelques familles de composés organiques ainsi que des principaux types de greffons des phases stationnaires actuelles.

Séparation de sucres sur une phase « amine ».
L’ordre d’élution des 7 sucres homologues nous indique que la phase stationnaire est polaire et que le pouvoir d’élution de la phase mobile diminue d’autant plus que la masse moléculaire est plus grande.

Effet de l’appariement d’ions sur une séparation avec une colonne type RP-18.
Agissant comme un savon, la partie hydrophobe de l’hexane sulfonate s’assemble, soit avec les chaînes C18 de la phase stationnaire dont la surface devient ainsi porteuse de charges anioniques, soit avec le soluté. La séparation d’espèces chargées s’en trouve améliorée.

Séparation sur le principe de l’interaction hydrophobe.
Chromatogramme d’un mélange de protéines, obtenu en diminuant au cours du temps la concentration saline. Les plus retenues sont les protéines les plus hydrophobes (d’après un document de la société Supelco).

Séparation sur le principe de l’interaction hydrophile (HILIC).
Chromatogrammes d’un mélange des 2 composés servant de marqueurs de temps mort sur une phase C18 (uracile, très polaire) et sur une phase HILIC (naphtalène, peu polaire) d’après un document de la société Macherey-Nagel. Noter l’inversion de l’ordre d’élution des 2 composés. H2O rappelle l’existence de la couche aqueuse à la surface de la phase stationnaire. En chromatographie de type HILIC, le toluène sert également de marqueur de temps mort.

Détection photométrique.
Principe d’un détecteur photométrique à une seule longueur d’onde et spectres d’absorption de quelques solvants utilisés en chromatographie liquide. On considère que la limite de transparence d’un solvant correspond à une absorbance de 0,2 pour 1 cm d’épaisseur traversée.

Chromatographie d’un échantillon contenant 2 composés A et B, dont les spectres UV sont différents.
Selon le choix de la longueur d’onde de détection, le chromatogramme n’aura pas le même aspect. Les chromatogrammes (à droite) d’un mélange de quelques pesticides enregistrés à trois longueurs d’onde différentes illustrent ce phénomène. En analyse quantitative, on doit donc déterminer d’abord les facteurs de réponse du détecteur pour tous les composés analysés (cf. analyse quantitative, chapitre 1).

Principe du détecteur à barrette de diodes.
À la différence du détecteur monochromatique, on est ici en présence d’un montage optique inverse, la cellule recevant à chaque instant toute la lumière issue de la source. Au fur et à mesure de l’élution, l’ordonnée du chromatogramme correspond à la somme des intensités envoyées par chaque diode (ex. : 1 024 diodes). Les données enregistrées permettent par la suite de calculer le spectre UV correspondant à chacun des analytes séparés.

Représentation sous l’aspect tridimensionnel, i = f(t, λ), d’une séparation chromatographique obtenue par une méthode d’enregistrement rapide (reproduit avec l’autorisation de la société TSP instruments).

Détection fluorimétrique.
1) Schéma de principe d’un détecteur fluorimétrique. 2) Chromatogramme d’un mélange d’hydrocarbures polynucléaires aromatiques (HPA). L’intensité de la fluorescence (390 nm) dépend de chaque composé, à la fois parce qu’elle dépend de la structure et de la longueur d’onde d’excitation choisie qui ne peut être optimale pour tous les composés. 3) Les amines primaires ou les aminoacides se prêtent à une transformation chimique pour les transformer en composés fluorescents (cf. chap. 11 § 6). Par réaction de l’orthophthalaldéhyde (OPA) puis du monothioglycol ou de l’acide mercapto-3 propionique (MPA), l’hétérocycle obtenu est fluorescent. Le MPA conduit à un composé plus polaire (R = CO2H) migrant plus rapidement sur une colonne de type RP. Cette dérivatisation peut être automatisée par les auto-échantillonneurs.

Un détecteur réfractométrique différentiel à déviation. Trajet optique au niveau de la cellule.
Le principe repose sur les lois de Fresnel de transmission de la lumière dans les milieux transparents dont l’indice de réfraction est n. Le contrôle de la position du faisceau réfracté est obtenu avec une photocellule à deux plages sensibles, dont on maintient les réponses égales par un coin optique (non représenté). Aspect d’un chromatogramme d’un mélange de sucres obtenu avec ce type de détecteur.

Détecteur évaporatif à diffusion de la lumière.
Par trois étapes successives, nébulisation, évaporation et détection lumineuse, un analyte présent dans la phase mobile est transformé en un aérosol apte à diffuser la lumière en provenance d’une source lumineuse. Les chromatogrammes représentés montrent que la sensibilité dépend fortement de la température de la zone chaude. La comparaison entre masse moléculaire et intensité des pics est ici très nette (d’après une notice d’application de la société Agilent).

Détection polarimétrique.
Schéma représentant les constituants principaux d’un détecteur polarimétrique avec, de manière superposée, l’effet sur la lumière entre la source et la photodiode détectrice. La lumière issue de la source, tout d’abord polarisée, traverse la micro-cellule de mesure placée en aval de la colonne puis un second polariseur qui servira d’analyseur dont l’axe de polarisation est croisé par rapport au premier assurant l’extinction du signal lumineux contrôlé par la photodiode. Au passage dans la cellule de mesure, l’angle de polarisation de la lumière va varier d’une valeur α. La présence d’un rotateur de Faraday dans le montage va permettre par induction d’un champ magnétique sur le matériau optique situé dans l’axe de la bobine de corriger cet angle a de façon à revenir à l’extinction du signal lumineux. C’est l’intensité de ce courant qui constitue le signal du détecteur (d’après le modèle OR-2090 de la société Jasco).

Chromatogrammes d’une séparation.
La phase mobile est un mélange binaire eau/acétonitrile : a) 50/50; b) 55/45; c) 60/40; d) 6/35. La flèche indique le temps mort tM (min), (selon J.W. Dolan, LC-GC Int, 1994, 7(6), 333).

Effet de la température de la colonne sur une séparation.
Exemple relatif à trois essais effectués sur un même mélange et avec un même débit de phase mobile à des températures différentes (a), 25 °C, (b) 35 °C et (c) 45 °C. Le temps mort quant à lui, est indépendant de la température.

Chromatographie rapide.
Cet exemple montre que le choix de la colonne et des conditions opératoires permet un gain de temps important sans altérer la résolution et l’efficacité (d’après un document de la société Shimadzu).

Comparaison des débits dans des colonnes de diamètres différents.
En admettant que le remplissage de ces quatre colonnes soit de même nature et de même porosité, on voit l’avantage que présente le passage de la CLHP classique (à gauche) à la CLHP-capillaire ou à la nano-CLHP (à droite). Plus le diamètre de la colonne est étroit, plus la consommation de phase mobile diminue, mais la reproductibilité des gradients est difficile à ce niveau.

Colonnes pour UHPLC. Nanochromatographie.
Colonnes capillaires de 75, 300 μm et de 2 mm de diamètre (Société Waters, colonnes Acclaim Pep Map). Exemple de séparation de 40 ng d’un mélange de protéines : ribonucléase, insuline, cytochrome C, myoglobine avec seulement 0,12 mL de phase mobile (document de la société Waters).







Montage théorique d’une installation de chromatographie par échange d’ions.
On retrouve une architecture modulaire qui se présente de manière intégrée ou non, avec, comme ici, deux modules optionnels (générateur d’éluant et suppresseur) et un détecteur basé sur la conductance des solutions. Le « suppresseur », intercalé entre la colonne et le détecteur, a pour rôle d’éliminer les ions de l’éluant par réaction de type acido-basique. Il est recommandé pour les phases mobiles très concentrées en ions.

Progression d’un anion A− au contact d’une phase stationnaire ammonium.
L’ion E− (généralement l’ion hydrogénocarbonate) est ici le contre-anion de la phase mobile. Il se fixe sur la phase stationnaire. L’anion A− (ion chlorure par exemple), qui est l’analyte, présent dans la phase mobile, vient prendre la place de l’ion E−. L’élution inverse le sens de la réaction d’équilibre en régénérant, au niveau du site considéré, l’état initial (fixation d’un ion E− ou d’un autre ion de même type). La progression de A− dans la colonne dépendra de son affinité avec les sites ioniques de la colonne.


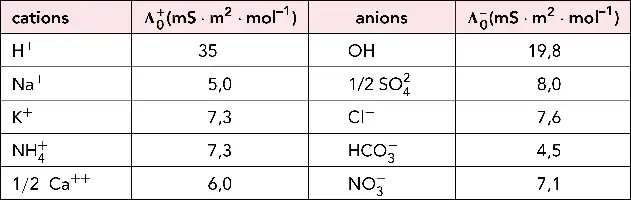
Phases stationnaires en CI.
Schéma d’une particule sphérique de polystyrène à usage d’échangeur de cations. Matrice de polystyrène transformée en résine échangeuse de cations (ex. DOWEX® 4 ou en résine échangeuse d’anions (ex. DOWEX® MSA-1, ou Permutite® si R = Me).

Phases anioniques obtenues par greffage d’une silice à noyau dur.
Gel de silice monodisperse non poreux.

Phases issues de polysaccharides.
Quelques exemples de résines commerciales obtenues à partir de la cellulose ou de l’agarose. Micrographie (50 000+) (d’après un document de la Société Interchim).

Générateur d’ion hydroxyde OH− à la demande.
Par électrolyse de l’eau, on génère au contact de la cathode des ions hydroxyde accompagnés de dihydrogène éliminé par un dégazeur à capillaire. KOH peut être remplacé par NaOH ou LiOH. Ce générateur est situé en amont de la boucle d’injection. (D’après un document de la société Dionex.)

Chromatogramme faisant apparaître le pic de l’eau (1 min) et le pic de système (16 min) et exemple de séparation d’anions.
L’acide phtalique (l’éluant) est un diacide organique (pKa1 = 2, 9 et pKa2 = 5, 5). La valeur du pH choisi étant intermédiaire (pH < 4, 5), il subsiste la forme non dissociée de ce diacide (d’après un document de la société Metrohm).

Suppresseur chimique à colonne de neutralisation.
Dans cet exemple de séparation de cations, le suppresseur chimique de type anionique purge l’éluant des ions H+ et de la quasi-totalité des ions Cl−, facilitant ainsi la détection du cation M+. Ce suppresseur contient une résine anionique (ex. \begin{equation}\mathrm{ArCH}_2(\mathrm{NR})_3^{+} \mathrm{OH}^{-}\end{equation}) qui va libérer des ions hydroxyle et ainsi neutraliser un nombre équivalent d’ions H+ pour former des molécules d’eau.

Principe de fonctionnement d’un suppresseur à membrane et d’un suppresseur à régénération électrolytique.
Il existe des membranes cationiques perméables aux cations (ici H+ et Na+), et anioniques perméables aux anions (ici OH− et Cl−). a) Modèle à membrane cationique adapté à l’élution des anions. La membrane consiste en une paroi polyanionique fixe repoussant par conséquent les anions de la solution ; b) Modèle électrolytique (électro-désionisation) comportant une membrane anionique, adapté par opposition au modèle précédent, à l’élution des cations. Il se régénère par électrolyse de l’eau. Noter, dans les deux cas, la circulation à contre-courant entre la phase éluée et la solution du suppresseur.

Chromatographie d’exclusion ionique.
Les ions, soumis à l’exclusion Donnan, restent dans le volume mort de la colonne alors que les acides faibles peuvent se fixer sur la résine, et même rentrer dans ses cavités. Ce mode de séparation des acides faibles peut être transposé aux sucres ou bases faibles avec une résine ammonium quaternaire et une phase mobile à base de soude diluée ou de carbonate de sodium. L’usage de colonnes spécialement adaptées à ce type de séparation est recommandé.

Analyse des acides aminés. Réaction d’échange sur la colonne et réactions de dérivation avec la ninhydrine.
Quel que soit l’acide aminé de départ, R désignant le reste de la molécule sous forme non-explicite, il se forme le violet de Ruhemann, dont le maximum d’absorption est à 570 nm. L’hydrindantine qui se forme par couplage de 2 molécules de ninhydrine est un catalyseur de la réaction. Une alternative consiste à transformer chaque amino-acide en un dérivé de l’indole fluorescent en présence d’aldéhyde o−phtalique et d’un dérivé d’un thiol.





Appareil de dépôt automatique en CCM et de lecture de plaque.
Applicateur Linomat IV programmable, et densitomètre mesurant la lumière réfléchie ou transmise par la plaque. Le schéma optique est assez semblable à celui d’un spectromètre UV/Visible (modèle Scanner 3, reproduit avec l’autorisation de la société Camag).

Chambre de développement à cuve verticale et plaque de CCM.
De dimensions variées en fonction de la taille des plaques (de 5 × 5 à 20 × 20 cm) elles sont en verre et munies d’un couvercle. Il existe d’autres cuves permettant une migration horizontale de la phase mobile. Aspect classique d’une plaque après révélation des spots de migration, calcul du Rf (cf. §4.4). x0 représente la distance de migration de la phase mobile.

En procédant à 2 élutions successives dans les 2 directions, on peut conclure que le composé X est un mélange d’au moins deux composés parmi lesquels le composé de référence a (même Rf dans les deux solvants), mais le second composé n’est pas b, bien qu’ayant le même Rf dans l’élution 2.

Couplage chromatographie couche mince/spectrométrie de masse (CCM/SM).
La plaque de CCM (1) après migration de l’analyte est placée dans un extracteur (2) qui permet de le récupérer par élution avec un solvant et de le transférer au SM pour y être analysé (3). Plusieurs sociétés commercialisent ces dispositifs (SM modèle Expression de la société Advion, sans sa console d’exploitation, mais avec son extracteur de plaques Plate express. Au centre, schéma d’un extracteur d’après un modèle de la société Camag).

Scannérisation d’une plaque de CCM.
L’absorbance d’une même plaque de CCM selon qu’elle est enregistrée à 254nm ou à 308nm montre que la sensibilité des mesures dépend du choix de la longueur d’onde (séparation de 5 sulfonamides, selon un document de la société Camag).

Micrographies de plaques de CCM et étude de la séparation de 4 parabènes sur des phases différentes.
a) À gauche, coupe d’une plaque CCM standard montrant les grains de gel de silice (10-15 μm) et à droite plaque lichrospher de Merck (diam. 8-10 μm). b) Sur phase polaire avec un éluant peu polaire l’ordre de migration (du plus petit déplacement vers le plus grand) correspond à l’ordre de polarité décroissante (le butylparabène le moins polaire migre plus vite). c) En revanche, sur phase inversée peu polaire, l’ordre de migration avec un éluant polaire correspond à l’ordre de polarité croissante.

Séparation de catécholamines en CCM avec la technique d’appariements d’ions.
On utilise ici une phase à polarité inversée et pour améliorer la séparation de ces composés polaires qui seraient insuffisamment retenus, on ajoute dans la PM un alkylsulfonate (cf. chap. 3 §7.1). Ce pseudochromatogramme a été obtenu par scannérisation de la plaque CCM (d’après une note d’application Merck).



Diagramme d’équilibre de phase du dioxyde de carbone.
Il existe pour chaque corps pur une relation entre les trois variables pression P, volume V et température T connue sous le nom d’équation d’état. Le diagramme ci-dessus est la projection (P/T) pour CO2. Le point critique est situé à 31°C et 7,4 MPa (1 MPa = 106 Pa, soit 10 bars). Il est possible de passer de l’état liquide à l’état gazeux en contournant le point critique, donc sans discontinuité de phase.

Moments dipolaires et échelle empirique de Snyder pour quelques modifiants.
Le modifiant permet d’agir de manière très fine sur les facteurs de rétention k des analytes, donc sur les sélectivités α. Selon la quantité de modifiants (entre 0 et 30 %), la pression en sortie ou la température, la force élutive de la PM sera modifiée. Pour cette raison on définit pour chaque modifiant une plage de polarités. Pour les composés très polaires, on ajoute au modifiant un additif tel le formiate ou l’acétate d’ammonium.

Schéma fonctionnel d’une installation de SFC pour colonne remplie de type CLHP.
Le dioxyde de carbone passe à l’état supercritique entre la pompe et l’injecteur. Un régulateur de pression (encore appelé restricteur) situé en aval de la colonne, avant ou après le détecteur, permet le maintien de la phase mobile à l’état supercritique jusqu’à l’extrémité de la colonne (dessin exécuté d’après un document de la société Vydac).

Comparaison entre CLHP et CPS.
Ces deux courbes expérimentales ont été obtenues avec la même colonne et le même composé, l’une en utilisant une phase liquide classique et l’autre du dioxyde de carbone à l’état supercritique. Les HEPT sont comparables — mais la séparation peut être conduite 3 fois plus rapidement en SFC, d’où un gain de temps.

Effet de la pression en SFC.
Ces deux chromatogrammes obtenus avec une colonne apolaire de type C18, montrent qu’en diminuant la pression de sortie au moyen du restricteur, la polarité de la PM diminue elle aussi, ce qui augmente les temps de rétention et le facteur de séparation (sélectivité). Le composé 2, moins polaire que le 1 est un peu plus retenu par la PS apolaire. On notera également la rapidité de l’analyse, moins de 50 secondes au total (d’après une étude de A. Morand, société Pharma-Physic).

Exemple de séparation sur colonne chirale.
Un mélange des 2 isomères cis/trans du nérolidol conduit à 4 pics dans les conditions indiquées (figure composée d’après un document de la société Waters). On remarquera ici encore que l’analyse ne dure que 3 minutes, alors qu’en CPG classique sur colonne capillaire cyclodextrine de 25 m il faudrait environ 30 minutes à 230°C. En dessous, structure moléculaire d’une phase à base d’amylose dérivée sous forme de carbamate, sur support de silice.

Comparaison de la CPS et des variantes de la CLHP.
Le domaine de la CPS recouvre l’éventail des variantes de la CLHP, tout en permettant des séparations plus rapides et une consommation moindre de phases mobiles.


Migration au travers du gel de phase stationnaire.
Chromatogramme figurant une séparation de trois espèces (1,2,3) de tailles différentes en solution. Les molécules les plus grosses 1 arrivent en tête, suivies des molécules moyennes 2 et enfin des petites molécules 3. Les volumes d’élution sont compris entre VI et VM = VP + VI. Image d’un gel poreux pour illustrer la notion de séparation selon la taille des pores.

Comparaison perméation de gel et filtration sur gel.
En utilisant une PS de perméation de gel à petits pores (5 nm), il est possible de séparer des molécules de faibles poids moléculaires (le pic du toluène permet de repérer Vm). Une colonne de filtration sur gel avec l’eau comme solvant est adaptée à la séparation de sucres ou de dextranes, macromolécules d’origine naturelle (Illustrations adaptées de documents de la société Varian Polymer Lab.)



Courbe d’étalonnage d’aspect linéaire d’une colonne de perméation de gel.
En utilisant deux mélanges complémentaires d’étalons de polystyrène on dispose d’un nombre suffisant de masses pour étalonner la colonne. La droite obtenue, qui recouvre un large domaine de masses (conséquence d’une phase stationnaire « panachée »), permet, dans un second temps de déterminer la masse moléculaire d’un polystyrène inconnu.

Courbe d’étalonnage universelle.
Figuration du rayon hydrodynamique d’un polymère dans un solvant non-miscible. Dans le cas d’une pelote polymère, ce rayon correspond au rayon d’une sphère dans laquelle le solvant ne pénètre pas.

Détection viscosimétrique.
Diagramme de principe d’un détecteur viscosimètrique. 4 capillaires sont répartis en 2 voies avec en plus un réservoir de phase mobile pure sur la voie dite de référence. La viscosité instantanée (et intrinsèque) est déduite des mesures de pression par les capteurs 1 et 2. Comparaison de deux chromatogrammes (détection réfractométrique et viscosimétrique) obtenus à partir d’un même mélange de polymères.

Diffusion à 90 degrés en prenant exemple avec une protéine.
L’association de deux détecteurs (indice de réfraction et diffusion sous un angle droit) permet d’observer la formation d’agrégats et d’en calculer les masses absolues, sans étalonnage préalable (d’après un document de la société Malvern). On remarque que le pic situé à 18mL correspond à une famille de masses variables, tandis que le pic à 25mL est apparemment formé par un composé de masse unique.

Cellule de mesure d’un détecteur multi-angle de lumière diffusée (MALS).
La cellule à circulation (40 μL) est constituée par un fin canal creusé dans un cylindre de quartz obturé par des fenêtres à ses extrémités. Le rayonnement laser traverse la cellule suivant son axe. Tout autour sont disposées des photodiodes qui récupèrent la lumière diffusée, chacune sous un angle précis. Chromatogramme d’un mélange de 2 protéines, avec en superposition le tracé des masses absolues calculé de manière répétitive tout au long de l’élution de chaque pic (d’après des documents de la soc. Wyatt).

Migration d’un mélange de deux macromolécules sur le principe du fractionnement par couplage flux-force (FFF)
Le champ transverse est ici produit par un gradient thermique obtenu en portant la paroi supérieure à une température plus élevée que la paroi d’accumulation. La séparation par FFF agit uniquement sur les propriétés hydrodynamiques et n’implique pas d’interactions avec une phase stationnaire.




Électrophorèse de zone : principe d’une installation.
A Chaque compartiment est séparé par un diaphragme afin d’éviter la contamination du tampon de migration par les produits secondaires qui se forment au contact des électrodes. On peut opérer soit à intensité, soit à tension, soit à puissance constante. B Après migration la bandelette peut faire l’objet d’un examen par densitométrie où les analytes sont « révélés » au moyen d’un réactif approprié. C Modèle à migration horizontale.

Électrophorèse capillaire.
L’électrolyte est une solution aqueuse ionique. L’échantillon est introduit dans la partie amont du capillaire (cf. § 3). Au-delà de 5-600 V/cm (ddp totale 30 kV si L = 50 cm), une isolation électrique devient nécessaire par sécurité. La distance utile de migration ℓ s’arrête au détecteur. Un électrophorégramme couramment effectué en laboratoire de biologie. Représentation en coupe d’un capillaire de dimensions courantes. Noter l’étroitesse du canal où circulent les solutions.

Déplacement des analytes dans le capillaire.
Influence de la charge nette, du champ électrique, du rayon hydrodynamique de l’ion et de la viscosité de la solution sur sa vitesse de migration dans un électrolyte (cf. § 2.2). L’électrophorégramme montre que les cations, les molécules neutres et les anions migrent dans la colonne en alimentation positive (champ électrique orienté de l’entrée vers la sortie du capillaire). La séparation dépend approximativement du rapport masse/charge de chaque espèce. Les espèces neutres ne sont pas séparées. Les anions de petite taille arrivent en dernier car sans le flux électro-osmotique ils se dirigeraient vers l’anode.

Flux électro-osmotique dans un capillaire rempli par un électrolyte.
Haut : Si la paroi de silice n’a subi aucun traitement (paroi polyanionique) il apparaît un transfert de l’électrolyte du compartiment anodique vers le compartiment cathodique : c’est le flux électro-osmotique. Il croît avec le potentiel appliqué et le pH (entre pH 7 et pH 8, υEOS peut augmenter de 35 %). Il est dû au potentiel existant à la surface de la paroi. Si on annule ce potentiel en recouvrant la paroi d’un film apolaire (film polyhydroxyvinylique hydrophile ou octadécyle, par ex.), ce flux disparaît. Bas : Modification dynamique de la paroi de silice par effet d’un surfactant cationique. Les migrations devant se faire vers l’extrémité où se trouve le détecteur, on doit inverser les pôles de l’appareil.

Électrophorègramme d’un mélange test d’anions.
Séparation d’un mélange d’anions avec la technique PAGE et détection par UV. D’après un document Beckman-Coulter.

Séparation des principaux acides organiques dans un vin blanc par détection inverse.
Les deux acides malique et lactique témoignent de la fermentation classique malo-lactique (selon une note d’application de la société TSP).

Un modèle d’interface entre ECHP et SM.
Les solutions diffèrent selon le mode d’ionisation choisi et selon qu’il y a appoint ou non d’un électrolyte additionnel. L’extrémité du capillaire est érodée pour le rendre perméable aux ions de l’électrolyte afin d’assurer la fermeture du circuit de l’ECHP. La pointe métallique du nébuliseur doit par contre être portée à un potentiel positif par rapport à l’entrée du spectromètre de masse.

Séparation d’analytes neutres par usage d’un surfactant (technique MEKC).
Dans cette illustration d’un capillaire non traité dans lequel a été ajouté au tampon de migration un surfactant polyanionique (par ex. le dodecylsulfate de sodium), on voit que les divers analytes sont retenus plus ou moins efficacement, par une suite d’équilibres réversibles (ici K2 > K1). Les micelles, polyanioniques, très lentement entraînées vers la cathode forment une phase quasi stationnaire. L’intérieur de chaque micelle est hydrophobe. L’électrophorégramme théorique traduit l’effet des interactions plus ou moins grandes entre les analytes et le surfactant.

Comparaison de la progression de la phase mobile en ECHP et en CLHP.
En ECHP, l’électrolyte et les analytes sont tractés par la paroi du capillaire alors qu’ils sont poussés en CLHP. La diffusion étant plus faible qu’en CLHP, l’efficacité (N) est donc très grande. Pour respecter un dessin fait à l’échelle, le diamètre de la colonne de CLHP devrait être au moins 20 fois plus grand que celui du capillaire de l’électrophorèse.

Séparation d’énantiomères.
Étude comparative entre ECHP et U-CLHP dans la séparation de 3 amphétamines racémiques (conc. 25 μg/mL) avec le même sélecteur chiral ajouté au tampon de séparation pour l’ECHP et à la phase mobile pour l’U-CLHP. L’électrophorèse permet une analyse rapide qui rivalise avec la chromatographie liquide la plus évoluée (d’après une étude de G. Bonvin, Thèse Université Genève 2014).

Séparation d’hydrocarbures aromatiques par électrochromatographie et représentation d’un segment de capillaire rempli.
Les différents essais de séparation d’un même mélange de composés neutres en faisant varier la tension vérifient, en première approximation, la relation existant entre tension et vitesse du flux électro-osmotique. Dans cet exemple, l’emploi d’une colonne très courte et d’une tension élevée permettent la séparation, en quelques secondes (pour V = 28 kV) avec une bonne résolution d’un mélange de composés non ionisés. On atteint une grande efficacité par ce procédé (D.S. Anex et Coll., Anal. Chem., 1998, 70, 4787-92). Parmi les matériaux de remplissage des capillaires, on trouve désormais des structures polymériques continues à base de polymères utilisant des méthacrylates ou des acrylamides.

Trois types de spectres rencontrés dans l’UV/Visible.
Spectres du benzène a) en solution (spectre de bandes) ; b) à l’état de vapeur (spectre présentant une structure fine) ; c) expansion d’une partie du spectre de la vapeur d’iode haute résolution (0, 14nm d’intervalle au total). Ce spectre s’étend sur 5 cm−1 soit de 536,83 à 536,97 nm.

Diagramme énergétique d’une molécule et transitions électroniques.
Sous la forme d’un diagramme d’état sont figurés, à gauche, 3 niveaux électroniques S avec quelques niveaux de vibration associés (on utilise souvent la lettre S pour désigner les états électroniques plutôt que E pour rappeler qu’il s’agit d’états singulets). Chaque trait horizontal correspond à un niveau d’énergie de la molécule. Au repos, seul l’état électronique fondamental S0 est occupé et l’état vibrationnel V0 est significativement le plus peuplé (conformément à la répartition de Boltzmann). Les transitions électroniques demandent environ 20 fois plus d’énergie que les transitions de vibration. Le passage de S0 à S2 correspond à une harmonique de la transition de S0 à S1. Au centre image d’un spectre UV qui montrerait la présence de 2 types de transitions, vers S1 et vers S2. À droite, représentation sous forme d’un diagramme énergétique, des différentes orbitales moléculaires dans la molécule de méthanal, au repos. Chacune contient 2 électrons aux spins appariés. Valeurs moyennes en nm des bandes d’absorption de cette molécule.

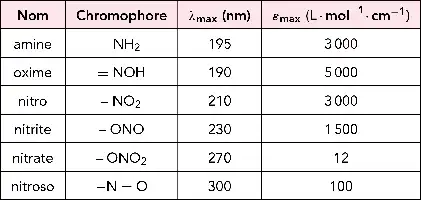
Les transitions les plus souvent rencontrées dans les composés organiques.
Parmi les quatre transitions réunies sur ce diagramme énergétique, les deux plus intenses correspondent à des transitions permises par la théorie des orbitales moléculaires, alors que les plus faibles sont dites « interdites ». Positions moyennes des bandes d’absorption correspondantes sur une échelle en longueurs d’onde.

Transition n → σ* de l’aniline (amine primaire).
La transition correspond à une augmentation du “poids” de la forme mésomère polaire. L’absorption de l’aniline correspondant à cette transition disparaît si on ajoute un équivalent d’un acide protonique type HX : il y a formation d’un sel d’ammonium qui mobilise le doublet libre de l’atome d’azote, nécessaire à cette transition.

Interaction donneur/accepteur.
L’absorption du complexe formé correspondant à hν3, est déplacée vers les grandes longueurs d’onde par rapport à hν1 et hν2. Sa position dépend du potentiel d’ionisation du donneur et de l’affinité électronique de l’accepteur. C’est une transition électronique entre OM d’atomes différents.

Valeurs des λmax d’une famille de polyènes E-disubstitués conjugués qui diffèrent par le nombre de doubles liaisons conjuguées. Cet effet est à l’origine de la couleur de nombreux composés naturels, tel le β–carotène “tout trans” (onze doubles liaisons conjuguées).

Spectres de la benzophénone dans le cyclohexane (---) et dans l’éthanol (—).
On observe ici avec deux solvants différents les effets bathochrome ou hypsochrome, sur les deux types de transitions.

Effet du pH sur l’absorption d’une solution de phénolphtaléine.
Ce composé est incolore pour des pH inférieurs à 8 et rose vif pour des pH supérieurs à 9,5. Le graphe représenté ici en trois dimensions montre bien que pour des pH acides il n’y a pas d’absorption dans la partie visible du spectre. En revanche, l’absorption vers 500nm qui apparaît quand le pH devient alcalin est responsable de la couleur bien connue de ce composé. On notera pour cet exemple, la modification des liaisons chimiques selon le pH.


Courbes d’émission d’une lampe Quartz Tungstène Halogène (QTH) et d’une source au deutérium.
L’échelle logarithmique rend compte des grandes différences d’irradiance selon les longueurs d’onde. En grisé, le domaine utile pour une source au deutérium. Vue générale et vue de dessus d’une lampe à deutérium. La lampe est amorcée avec une tension de 300 à 400 V. La cathode est un filament d’oxydes métalliques émetteur d’électrons, alimenté sous quelques volts. Les pics d’émission du deutérium à 486,0 et 656,1 nm sont souvent utilisés pour régler les spectromètres.

Monochromateurs à réseau.
a) Montage d’Ebert comportant un miroir sphérique concave M3 et un réseau en configuration déportée. Il donne une excellente qualité d’image en compensant les aberrations ; b) montage Czerny-Turner, de conception voisine, comportant deux miroirs sphériques M3 et M4 et 2 réseaux R1 et R2 pour une meilleure résolution spectrale ; c) montage avec un réseau concave RC permettant à la fois dispersion et focalisation du rayonnement.

Courbes de réponses de quelques détecteurs utilisés dans la gamme 200 – 3 000 nm.
Photomultiplicateur (PMT), silicium (Si), sulfure de plomb (PbS), arséniure de Gallium-Indium (pour les mesures dans le proche infrarouge) et CCD au silicium. À droite, photodiodes d’une barrette CCD utilisées comme pixels en mode photampérique.

Schéma optique simplifié d’un spectrophotomètre simple faisceau de mode séquentiel.
1 : Deux sources coexistent mais une seule est choisie en fonction de la mesure. 2 : le monochromateur sélectionne la longueur d’onde de mesure. 3 : compartiment de mesure où une cellule contenant soit l’échantillon soit un blanc est placée sur le trajet optique. 4-5 : diode détectrice et diode de contrôle.

Spectrophotomètres simple faisceau à détection par barette CCD.
1) Composition du modèle UV7 "Fast Touch" (Mettler Toledo). 2) Agencement sous forme de modules d’un appareil portable (Ocean Optics) qui réunit une source (tungstène ou xénon), un spectrographe à barrette de diodes et une cellule insérée sur le parcours d’une fibre optique pour mesurer l’absorbance à distance. De nombreuses réalisations de ce type sont utilisées pour des applications de routine.

Parcours optique de deux appareils à double faisceau, entre la sortie du monochromateur et le détecteur (modèle à miroirs tournants et modèle à miroir semi-transparent).
L’agencement des appareils à miroirs tournants est à rapprocher de celui des spectrophotomètres IR, hormis le fait que la lumière issue de la source passe d’abord par le monochromateur avant d’arriver sur l’échantillon. On minimise ainsi les réactions de photolyse qui pourraient survenir par exposition prolongée à la totalité des radiations de la source. L’optique plus compacte des montages à un seul faisceau divisé associé à deux détecteurs est plus simple. Un miroir semi-transparent et fixe remplace le mécanisme délicat des miroirs tournants synchrones.

Cellules et principaux dispositifs d’échantillonnage.
a) cuve standard et cuve à circulation pour l’étude des solutions ; b) modèle de sondes à immersion. Extrémités d’une sonde classique et d’une sonde de type ATR. La lumière monochromatique issue du spectrophotomètre est guidée vers une cellule à immersion puis retourne vers le détecteur. Le trajet de référence est également assuré par une autre fibre optique. Le prisme saphir a un indice plus grand que celui de la solution ; c) courbe de transmittance de quatre matériaux utilisés dans le domaine de l’UV-Visible en fonction de la longueur d’onde.

Absorption de la lumière par un matériau homogène, et représentation du % de transmission en fonction de l’épaisseur traversée.
La lumière arrivant sur un échantillon peut être transmise, réfractée, réfléchie, diffusée ou absorbée. Ici on ne prend en compte que la partie absorbée.

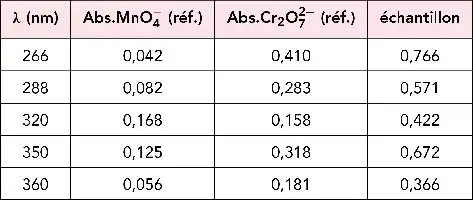
Illustration de la loi de Beer et Lambert.
Spectres de solutions aqueuses de concentrations croissantes en permanganate de potassium. Graphe des absorbances correspondantes mesurées à 525 nm montrant la croissance linéaire de ce paramètre.

Additivité des absorbances.
Pour toute longueur d’onde, l’absorbance d’un mélange est égale à la somme des absorbances de chaque composant du mélange (pris à la même concentration) pour cette longueur d’onde.

Point isobestique.
Hydrolyse alcaline à 25 °C du salicylate de méthyle. Superposition des spectres successifs enregistrés entre 280 et 350 nm à des intervalles de 10 min.

Illustration de deux situations fréquemment rencontrées.
Un composé masqué dans un mélange ou bien ne présentant pas d’absorption nette peut néanmoins être dosé par colorimétrie en faisant appel à une réaction chimique qui le transforme en un dérivé absorbant exempt d’interférences.

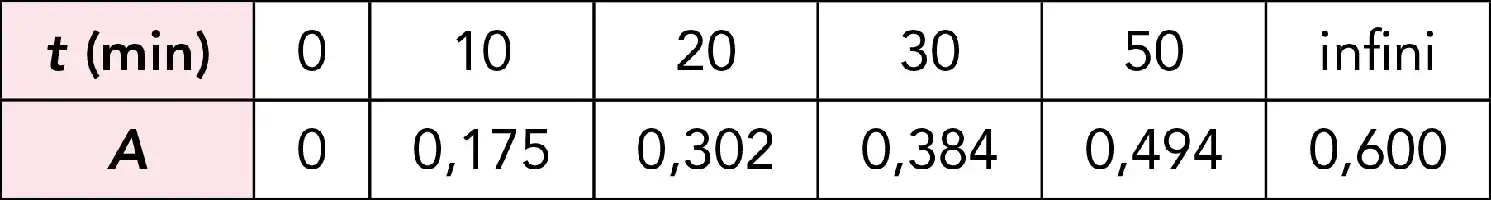
Courbe d’étalonnage.
Si on se contente de préparer une seule solution de référence, ceci revient à considérer que la courbe d’étalonnage est une droite qui passe par l’origine. La précision du résultat sera d’autant meilleure que la concentration inconnue sera proche de la concentration de référence (le résultat est déterminé par interpolation).

Analyse de confirmation.
a) Spectre d’un mélange (X + Y) et spectre de Y seul dans le même domaine (partie grisée) ; b) dessin illustrant un chromatogramme qui présente deux pics dont le premier est dû à un composé unique et le second à l’élution de deux composés légèrement décalés dans le temps. L’évolution du rapport des absorbances en cours d’élution permet de contrôler la pureté des composés élués. Ces variations sont habituellement mises en évidence par les logiciels sous forme de plages colorées.

Analyse multicomposants.
Spectres d’une solution 1 × 10−4 M en permanganate, d’une solution 1 × 10−4 M de bichromate et d’une solution contenant 0,8 × 10−4 M de permanganate et 1,8 × 10−4 M de bichromate (d’après Bianco et Coll., J. Chem Educ., 1994, 66(2), p. 178).

Déconvolution du spectre d’un mélange de 5 composés.
À partir du spectre expérimental du mélange, le logiciel retrouve la proportion de chacun d’eux (dont les spectres individuels sont connus par ailleurs).

Illustration se rapportant aux calculs de la méthode de Morton et Stubbs.
a - Spectre de l’échantillon. b - Spectre de l’analyte pur.

Courbes représentatives moyennes de chacune des causes d’erreur dans la mesure de l’absorbance (1, 2, 3), ainsi que la courbe globale résultant de leur somme (4) (voir texte).

Courbes dérivées de deux composés.
On peut noter la présence d’un minimum de la dérivée d’ordre 2 pour la position d’un maximum pour le spectre d’ordre 0, alors que pour la dérivée d’ordre 4, on retrouve sensiblement une inversion du tracé d’ordre 2 (figure composée à partir d’un document de Joanna Karpinska (2012). Basic Principles and Analytical Application of Derivative Spectrophotometry, ISBN : 978-953-51-0664-7).

Effet de la diffusion lumineuse sur un spectre UV et sur sa courbe dérivée.
Comparaison des courbes dérivées correspondant pour A au spectre d’un composé en solution sans phénomène de diffusion et pour B au même spectre en présence de diffusion. On observe que la diffusion perturbe ici de 10% l’absorbance mais de seulement 1% environ la valeur de l’amplitude de la courbe dérivée (spectres modélisés à partir de fonctions correspondant à des courbes de Gauss).

Colorimétrie visuelle.
Comparateur visuel (Société Merck) comportant deux tubes : l’un pour l’échantillon, l’autre pour le blanc. Devant ce dernier, on fait tourner un disque représentant l’évolution de la teinte en fonction de la concentration en analyte jusqu’à ce que les deux fenêtres aient la même teinte. Le colorimètre de Duboscq a été utilisé jusqu’au milieu du siècle dernier. Gràce à un système de prismes, il permet de comparer dans le champ visuel les intensités lumineuses ayant traversé deux cuves dont l’une contient l’échantillon à quantifier et l’autre un témoin de référence. Ce dispositif original conduit à la concentration cherchée en exploitant la loi de Beer et Lambert par comparaison des épaisseurs taversées.

Interprétation « mécanique » de l’interaction d’une onde lumineuse avec une liaison polarisée.
La fréquence mécanique de l’onde n’est pas changée par absorption du photon, seule son amplitude croît.

Spectre d’un film de polystyrène dans le moyen infrarouge.
Représentation habituelle d’un spectre, avec en abscisse une échelle en cm−1 (voir formule 10.1), linéaire sur toute l’étendue spectrale, et en % de transmittance en ordonnée (pour une observation plus aisée de la partie droite du spectre, on fait assez souvent un changement d’échelle à 2 000 cm−1 (voir figure 10.9). La transmittance est quelquefois remplacée par l’absorbance A (A = −log T). L’échelle, en cm−1, (ou kaysers) est linéaire en énergie (E = hc/λ), et va en diminuant de la gauche vers la droite (des fortes aux faibles énergies).

Une molécule diatomique représentée sous la forme d’un oscillateur harmonique.
Le terme d’oscillateur harmonique, a pour origine la relation directe entre l’élongation et la force exercée, alors que la fréquence νVib. en est indépendante.

Diagramme des énergies de vibration d’une liaison.
La transition V = 0 à V = 2 correspond à une bande harmonique de faible intensité. Compte tenu des énergies des photons dans le moyen infrarouge, on peut calculer que le premier état excité (V = 1) est 106 fois moins peuplé que l’état fondamental. Les transitions harmoniques sont exploitées dans le proche infrarouge.

Niveaux de rotation/vibration d’une molécule diatomique et spectre correspondant. Bande de vibration rotation du monoxyde de carbone.
En appelant V et J les nombres quantiques de vibration et de rotation, la vibration fondamentale correspond à ΔV = +1 et ΔJ = +1. a) Une bande de vibration-rotation correspond à tous les sauts quantiques autorisés. Si l’échelle du diagramme est en cm−1, les flèches correspondent aux nombres d’onde des absorptions ; b) la branche R correspond à ΔJ = +1, et la branche P à ΔJ = −1. Elles sont situées de part et d’autre de la bande Q (transition ΔJ = 0), absente car interdite pour les molécules diatomiques ; c) Bande de vibration-rotation du monoxyde de carbone (P = 1 000 Pa). Les nombreuses raies observées sont en accord avec les règles de sélection. L’intervalle entre les raies successives de rotation varie progressivement, par suite du facteur d’anharmonicité.

Vibrations moléculaires du CH2.
Vibrations caractéristiques d’élongation et de déformation, dans le plan et hors du plan (oop, « out of plane »). Dans l’infrarouge, la position et l’intensité des bandes sont modifiées par les associations entre molécules, la polarité des solvants, etc.

Configurations des spectromètres et analyseurs dans l’infrarouge.
a) Spectromètre simple faisceau à transformée de Fourier ; b) analyseur simple faisceau, comportant un monochromateur fixe ou un filtre, utilisé lorsqu’une mesure à longueur d’onde unique suffit ; c) spectromètre double faisceau de type dispersif. Contrairement aux spectrophotomètres de l’UV/Visible, l’échantillon, placé avant le monochromateur, est soumis en permanence à tout le rayonnement de la source. L’énergie des photons dans ce domaine est insuffisante pour briser les liaisons et, par cela même, dégrader l’échantillon.

Montage optique d’un appareil à transformée de Fourier.
a) Interféromètre de Michelson 90° avec, en cartouche, quelques détails au niveau de la séparatrice ; b) diagramme optique d’un spectrophotomètre à simple faisceau (dessin du modèle 8 300 de la société Shimadzu). Un laser He/Ne de faible puissance émettant dans le visible (632,8nm) est utilisé comme étalon interne afin de repérer avec précision la position du miroir mobile par une méthode interférentielle (ce second interférogramme sinusoïdal suit le même trajet optique).

Séquence d’obtention d’un spectre pseudo double-faisceau avec un spectromètre infrarouge à transformée de Fourier.
L’appareil enregistre et met en mémoire deux spectres qui représentent les variations de I0 (le blanc) et de I (l’échantillon) en fonction du nombre d’onde (ce sont les spectres en émittance 1 et 2) ; puis il calcule le spectre conventionnel, identique à celui que donne un appareil de type double faisceau, en effectuant le rapport T = I/I0, pour chaque nombre d’onde. L’absorption atmosphérique de CO2 et H2O (bandes d’élongation et de déformation observées en 1 sur le spectre sans composé) se trouve ainsi éliminée. Les illustrations correspondent à la réalisation d’un spectre d’un film de polystyrène.

Analyseur IRTF/Raman portable.
Cet appareil de terrain intègre les 2 techniques, IRTF et Raman, pour l’analyse des liquides et des poudres. À la partie supérieure on distingue le dispositif ATR simple réflexion (cf. § 9.3)pour les analyses par infrarouge et l’emplacement destiné au flacon échantillon pour les analyses par spectrométrie Raman. L’instrument dispose d’une spectrothèque intégrée pour une identification immédiate de l’échantillon (modèle Gemini de la société Thermo-Scientific).

Analyseur de gaz de type non dispersif.
La cellule contenant le gaz à quantifier est traversée alternativement par deux faisceaux de longueurs d’onde différentes (miroir semi-transparent) définis par le choix de deux filtres, l’un (F1) situé dans une zone sans absorption par le gaz (voie de référence) et l’autre (F2) correspondant à une longueur d’onde spécifique du gaz. L’absorbance obtenue à partir des deux mesures conduit à la concentration cherchée. Illustration avec le spectre du dioxyde de carbone sur lequel sont superposées les bandes passantes des deux filtres F1 et F2.

Quelques sources utilisées dans le proche et moyen infrarouge.
En haut : quelques sources proposées pour équiper les instruments conventionnels ou particuliers (sociétés Oriel (1), Hawkeye (3), Thorlab (4), Axetris (6)).En bas : source de faible puissance (0,5 W), destinée aux analyseurs infrarouges portables (EMIRS-50 de la société Axetris). Empruntant la technologie des systèmes micro-usinés. Fonctionnant à moins de 1 000 °C, la courbe d’émission est quasi parfaite comparée à celle d’un corps noir à la même température. Ces sources avec ou sans réflecteur peuvent être protégées par une fenêtre d’un matériau transparent dans l’IR (ex. BaF2).

Détecteurs dans l’infrarouge.
Principe de fonctionnement des détecteurs pyroélectrique et à semi-conducteur. Au centre, aspect général du détecteur et de son boîtier. En bas, plage d’utilisation des principaux détecteurs.

Cellules dans le moyen infrarouge.
Vues d’une cellule à gaz à trajet optique direct (10 cm) et d’une cellule de conception classique pour films liquides (d’après des documents de la société SpectraTech). Ce type de cellule démontable destiné à des analyses qualitatives, permet de changer rapidement la nature des fenêtres.

Matériaux et solvants dans le MIR.
Les principaux cristaux ou verres utilisés pour réaliser les parois des cellules, sont utilisables en deçà des limites indiquées. Quelques composés à usage de solvants dans les domaines indiqués (zones roses).

Angle critique et onde évanescente.
Comparaison du parcours, réfléchi ou réfracté, de deux rayons suivant leur angle d’incidence. Dispositif par réflexion totale atténuée (ATR) avec un cristal pyramidal qui ne permet qu’une seule réflexion. Réalisation commerciale de ce type (Specac Ltd.). Le contact entre l’échantillon et le cristal protégé par une lamelle en diamant (n = 2,42), est facilité par écrasement.

Les 3 types de réflexions utilisées dans le MIR.
a) Dispositif par réflexion spéculaire. Parcours optique dans un dispositif à angle fixe de 30° pour les échantillons très réfléchissants et autre montage à 8 degrés b) Dispositif par réflexion totale atténuée (ATR) par emploi d’un cristal pyramidal pour réflexions multiples. c) Schéma optique d’un dispositif pour réflexion diffuse et dessin à vue d’une réalisation Spectra-Tech.

Spectres en réflexion.
a) Spectres d’un échantillon de plexiglas obtenus par utilisation des trois dispositifs de réflexion. À gauche les spectres bruts et à droite les spectres après correction. En haut : signal brut de réflexion spéculaire et le résultat en unités « K » par application du calcul de Kramers-Kronig (transformation de la réflectance) ; au milieu, spectre obtenu en lumière diffuse : comparaison du spectre brut et du spectre corrigé selon Kubelka-Munk ; en bas spectre obtenu par ATR, ce dernier nécessite une correction peu importante diminuant l’absorbance pour les grandes longueurs d’onde qui serait surévaluée ; b) Comparaison de 2 spectres de l’acide benzoïque, l’un par transmission, l’autre par réflexion diffuse et correction K-M.

Trajet optique dans un microscope IR.
L’échantillon peut être observé en transmission (dessin de gauche), ou en réflexion spéculaire (dessin de droite). Cet accessoire s’installe sur un spectromètre dont le faisceau est dévié. L’optique Cassegrain qui est appropriée pour l’examen des petits objets a aussi pour avantage dans l’IR que la lumière est réfléchie à la surface de miroirs, sans avoir à traverser des lentilles optiques.

Calcul de l’épaisseur d’une cellule par la méthode des franges d’interférences.
À gauche, réflexions sur les parois d’une cuve (pour plus de clarté, l’angle d’incidence du faisceau sur la cellule est décalé de l’incidence normale d’un petit angle ; à droite, partie d’un enregistrement obtenu à partir d’une cellule vide. On dénombre 12 franges entre les deux flèches. Le calcul (expression 10.11) conduit ici à ℓ = 204 μm.

Correction du fond d’absorption.
En supposant, que dans l’exemple reproduit ci-contre, la mesure de concentration soit basée sur l’absorbance de la bande à 1070 cm−1, il faut retrancher à l’absorbance totale (0,29), le fond d’absorption de 0,05, ce qui conduit à 0,24 u-abs.


Effet de la granulométrie sur le spectre d’une farine et courbes dérivées.
En haut : Comparaison des spectres d’une même farine selon sa granulométrie (Osborne et al.) A, mouture grossière, B, mouture fine. Si la mesure était effectuée à une seule longueur d’onde (ex. 1 940 nm), il semblerait que le contenu en protéines soit différent, mais si on se réfère à l’ensemble du spectre, on voit qu’il s’agit du même produit. En bas : Comparaison des spectres de dérivée seconde dans le proche infrarouge de l’eau d’une part et d’un mélange moitié eau/moitié méthanol. On remarque qu’il est possible de doser le méthanol à partir des courbes sigmoïdales qui se situent vers 2 300 nm, sans interférence avec le signal de l’eau observé vers 1 440 et 1 940 nm.

Sphère intégrante et réalisations commerciales en SPIR.
À gauche, schéma d’une sphère d’intégration (DIAM qq. cm) à une seule voie, utilisable sur les spectromètres IRTF. La paroi interne réfléchissante (or ou téflon) permet aux multiples réflexions de générer une luminance uniformément diffusée quel que soit l’angle d’observation, un déflecteur protégeant le détecteur (ex. InGaAs) afin qu’il ne reçoive pas de lumière directe venant de l’échantillon. À droite, sphère intégrante pour appareil séquentiel (d’après document Shimadzu, modèle ISR-603). Spectrophotomètre Nicolet iS-10 (Thermo Scientific) équipé d’une sphère intégrante et spectromètre on line en temps réel Prediktor (Spektron), 960-1 700 nm (la source lumineuse est extérieure à l’appareil).

Spectromètres portables du proche infrarouge.
À gauche, spectromètre du proche infrarouge pour l’analyse des produits laitiers et des céréales (modèle StellarCase de la soc. StellarNet Inc.). À droite, appareil fonctionnant en réflectance diffuse pour l’analyse de matières premières (modèle QualitySpec Trex de la soc. Malvern Pananalytical). Ces deux appareils fonctionnent suivant le principe de la réflectance diffuse.

Diagramme énergétique et diffusion Raman.
L’utilisation d’un diagramme énergétique, de type Jablonski, pour représenter l’évolution de l’énergie d’une liaison stimulée par laser rend compte du phénomène de diffusion Raman et permet d’insérer les niveaux d’énergie virtuels. Une molécule polyatomique possède de nombreux degrés de liberté dont certains sont actifs en infrarouge (si le moment dipolaire varie) et d’autres en Raman (si la polarisabilité varie).

Spectre Raman.
a) Les trois principales raies Stokes et anti-Stokes de la molécule de tétrachlorure de carbone (excitation : laser argon ionisé, 488 nm) ; b) spectre Raman du même composé ; c) spectre réel de la « raie » 1333 cm−1 du diamant. Par utilisation d’une plus grande longueur d’onde, le spectre Raman est obtenu sans fluorescence gênante.

Spectre Raman d’un film de polystyrène réticulé.
Comparaison avec le spectre infrarouge, en noir sur la figure, enregistré en transmittance dans le même domaine spectral.

Spectromètre miniaturisé Raman.
Modèle BRAVO. Exemples de spectres obtenus avec cet appareil (gamme 300 – 3 200 cm−1) (Reproduit avec l’autorisation de la société Bruker).

Comparaison des intensités d’absorption (IR) et de diffusion (Raman) pour quelques liaisons chimiques.

Correlation dans le moyen infrarouge entre groupes fonctionnels et bandes d’absorption.



Représentation sous forme d’un diagramme énergétique, de l’absorption d’un photon par une molécule.
Transfert d’un électron d’une orbitale occupée (HO) vers une orbitale vacante (BV) avec apparition d’un état singulet évoluant en un état triplet, plus stable (transition n → π* par ex.). Les petites flèches désignent un électron dont le spin est figuré par le sens de la flèche.

Diagramme de Jablonski.
Selon la théorie quantique, la fluorescence résulte de transferts entre états de spin de même multiplicité, et la phosphorescence entre états de multiplicités différentes. L’état T1 produit un retard dans le retour à l’état fondamental pouvant atteindre plusieurs heures. Le « Stokes shift » correspond à l’énergie dissipée sous forme de chaleur (relaxation vibrationnelle) pendant la durée de vie de l’état excité, donc avant que les photons ne soient émis. Cependant la situation réelle est plus complexe que ce diagramme très simplifié peut le laisser supposer. À l’échelle macroscopique, un composé peut être à la fois fluorescent et phosphorescent car à l’échelle moléculaire les espèces individuelles qui le composent n’ont pas toutes le même comportement. Les flèches ondulées correspondent à des relaxations vibrationnelles sans émission de photons.

Représentation sur un même graphe des spectres d’absorption et de fluorescence du divinylcyclopentène.
Le spectre de fluorescence qui ressemble à l’image dans un miroir du spectre d’absorption, rend compte du « déplacement de Stokes » (Stokes shift) » qui trouve une explication en considérant les diagrammes énergétiques (fig 11.2) Exemple extrait de H. Jacobs et Coll, Tetrahedron1993, p. 6 045.

Composés fluorescents aromatiques.
Le nom est suivi du rendement de fluorescence Φf (voir § 11.3)dont la valeur est obtenue de proche en proche par comparaison avec des composés de fluorescence connue. Les mesures sont faites à 77 K. La 8-hydroxyquinoléine est représentative de diverses molécules formant des complexes de chélation fluorescents avec certains ions métalliques.

Intensité de fluorescence.
Suivant l’endroit de la solution où la fluorescence est émise, une intensité lumineuse variable atteint le détecteur ; il est possible d’évaluer la réabsorption de la lumière de fluorescence (comparaison entre a et c) et l’absorption de la lumière incidente (comparaison entre a et b). Par la présence d’un iris, seule la lumière, en provenance de la partie centrale de la cuve, est recueillie.

Intensité de fluorescence et concentration.
Modélisation à partir de la formule (11.7) de l’effet de la concentration sur l’intensité de fluorescence. On observe un maximum de fluorescence au-delà duquel elle diminue. Plus la solution est concentrée, plus faible est la fluorescence — sorte de roll over ou de self quenching.L’illustration correspond à 3 enregistrements à même échelle de solutions de biacétyle dans le tétrachlorométhane. La courbe correspond à la lumière recueillie en provenance du petit volume de côté ℓ situé au sein de la solution comme précisé sur la figure 11.4 (paramètres ℓ et ε choisis arbitrairement).

Les diverses composantes d’un spectre de fluorescence.
La position de la raie Raman dépend de la longueur d’onde de la raie excitatrice et du solvant. À droite, test de sensibilité d’un fluorimètre.

Positions des pics Raman, calculées pour quatre solvants usuels et cinq longueurs d’onde excitatrices d’une lampe à mercure

Agencement des différentes composantes d’un spectrofluorimètre et lampe à arc xénon.
La fluorescence est mesurée en régime permanent, en maintenant l’excitation, à la différence de l’étude de la fluorescence dynamique. Micro-cuve à circulation et cuve standard à géométrie 90 degrés. Modèle de lampe à arc xénon. La pression de xénon dans la lampe est d’environ 1 MPa. Ces lampes à arc (sans filament), à enveloppe en verre de silice, sont des sources de « lumière blanche ». La cathode correspond à l’électrode la plus fine.

Schémas optiques simplifiés de lecteurs de fluorescence pour micropuits.
À gauche, modèle à deux fibres optiques pour amener la lumière excitatrice au niveau du puits choisi et pour récupérer la lumière émise par l’échantillon, ici sous une géométrie frontale. À droite un modèle (Synergy-2 de BioTek) comportant en plus un miroir dichroïque afin d’améliorer la sensibilité.

Schéma du spectrofluorimètre Shimadzu F-4500.
Une fraction du faisceau incident, réfléchie par un miroir semi-transparent, arrive sur une photodiode de référence. La comparaison des signaux des deux détecteurs permet de compenser la dérive de la source. Ce procédé à un seul faisceau permet d’obtenir la stabilité propre aux appareils à double faisceau. Les spectres présentent souvent des petites différences lorsqu’ils proviennent d’appareils différents (reproduit avec l’autorisation de la société Shimadzu).

Spectres de fluorescence.
En haut : Matrice d’émission-excitation d’un mélange de deux ions fluorescents. Ce type d’enregistrement permet de trouver la meilleure longueur d’onde d’excitation. À droite, 1 correspond à un spectre de fluorescence (excitation λ1) et 2 à la fluorescence observée à λ2 quand on fait varier la longueur d’onde excitatrice. En bas : Exemple de spectres d’émission-excitation A et B.

Représentation de la décroissance de fluorescence et principe de la mesure.
Sur le graphe, chaque point figure le contenu d’un canal mémoire (couple temps/nombre de photons). La courbe de décroissance exponentielle attendue apparaît ici sous forme d’un tracé linéaire, ce qui est dû au choix de l’échelle logarithmique pour les ordonnées. L’illustration de droite indique comment on accède à cette représentation.

Processus d’un dosage de protéine par chimifluorescence en biochimie.
La figure regroupe simplement les réactions mises en jeu sachant que pour réaliser un tel dosage on suit un protocole dans lequel les différentes réactions se font au cours d’étapes distinctes.

Comparaison d’une détection UV et par fluorescence après séparation chromatographique.
Les aflatoxines, contaminants cancérogènes présents dans certains lots de céréales, font l’objet de contrôles par CLHP. On remarquera que, par détection UV, les intensités des pics varient comme les concentrations des 4 composés, alors que la détection par fluorescence est beaucoup plus sensible pour les G2 et B2 (reproduit avec l’autorisation de la société SUPELCO). En bas à gauche, agencement des différentes parties d’un détecteur basé sur la fluorescence. Il permet de trouver pour chaque composé élué le meilleur couple excitation/émission en un temps très bref sans interrompre le déroulement de la chromatographie. Les chromatogrammes obtenus sont étudiés en différé (reproduit d’après un document de la société Agilent technologies).

Exemples de réactions de chimiluminescence.
La nature des produits de réaction est quelquefois mal connue. Le luminol émet une intense lumière « bleu électrique ». Quant à l’oxalate de diphényle il permet d’obtenir, suivant le colorant utilisé, des émissions dans des couleurs très variées. Le dérivé aromatique à l’usage de « colorant » représenté est utilisé pour générer la couleur bleue des bâtons lumineux.

Analyseurs d’azote par chimiluminescence.
Réaction stœchiométrique de l’ozone sur le monoxyde d’azote. Moyennant la combustion de l’échantillon contenant l’élément azote, ou la réduction catalytique de NOx (ex. NO2) en NO (utilisation d’un convertisseur catalytique dédié) cette réaction de luminescence est exploitée dans de nombreux appareils de mesure (luminomètres), dont certains donnent une réponse quasi instantanée (contrôle des émissions automobiles). La mesure de l’azote total permet de remplacer la méthode de Kjeldahl (cf. chap. 21 § 8).

Réactions de chimiluminescence faisant intervenir l’ozone.
La transformation d’un composé contenant l’élément soufre en dioxyde de soufre à l’état excité, par l’ozone est réalisée dans plusieurs instruments dédiés à ce dosage. La dernière réaction peut s’appliquer au dosage de l’ozone comme de l’éthylène.


Fluorescence X, émission Auger et diffusion Compton.
Fluorescence X et émission Auger : le rayonnement ionisant éjecte un électron interne de l’atome, ce qui crée une lacune qui est comblée par un électron plus externe (de plus grande énergie). L’énergie excédentaire se retrouve, soit sous forme d’un photon émis (fluorescence X), soit éliminée sans radiation, par éjection d’un électron de l’atome (émission Auger). Diffusion Compton : le photon X incident éjecte un électron périphérique («électron de recul »). Le photon incident est d’autant plus dévié que son énergie est plus faible.

Schéma simplifié montrant l’origine de quelques transitions de fluorescence.
Sur l’image classique d’un atome de masse atomique moyenne, sont représentées les différentes réorganisations des électrons suite au départ d’un électron de la couche K. Ces transitions sont très peu affectées par la nature de la combinaison chimique dans laquelle se trouve l’atome. Ainsi, la raie Kα1 du soufre passe de 0,5348nm pour SVI à 0,5350 nm pour S0, soit un écart d’environ 1 eV, comparable à la largeur naturelle des raies X.

Générateurs de rayons X.
a, Schéma d’un tube classique à rayons X avec refroidissement par eau, indispensable si le tube est de forte puissance (1-4 kW), b, spectre émis par l’anode d’un tube à rayons X. On distingue le spectre continu dont l’étendue dépend de la tension appliquée et le spectre de raies de l’anode. Pour constituer des sources monochromatiques, on a recours à des filtres dont le spectre d’absorption et l’épaisseur isolent une seule raie (courbe en pointillés). c, modèle de générateur miniaturisé (Soc. Amptek). d, Générateur utilisant un cristal pyroélectrique, de très faible encombrement (diam. 15 mm). La production de RX correspond sur le dessin à la phase de refroidissement du cristal (raies du Cu). Le cycle chauffage/refroidissement dure environ 3 minutes e, modèle de ce type (Cool-X de la Soc. Amptek).

Source radioactive 55Fe.
Enregistrement du spectre d’émission d’une source de 55Fe obtenu en plaçant cette source dans le compartiment échantillon d’un spectromètre à dispersion en énergie. Les signaux correspondent à la fluorescence X du 55Mn, c’est-à-dire au noyau fils du 55Fe. La résolution de ce spectre, mesurée à mi-hauteur du pic principal est d’environ 136 eV.


Poire d’interaction d’un faisceau d’électrons avec un matériau.
Différents phénomènes se produisent au sein du matériau. Il en résulte une émission complexe dans laquelle on peut différencier l’origine des divers rayonnements recueillis. Si l’énergie est élevée, les rayons X sont formés plus profondément dans la matière. Il sera donc plus difficile de les détecter.

Différents détecteurs utilisés en spectrométrie de fluorescence X.
a, compteur en mode proportionnel. b détecteur à scintillation (cristal et tube photomultiplicateur). c transducteur à semi-conducteur avec une importante zone active. d, compteur à impulsion, du type SDD dont la série des électrodes annulaires crée un champ transversal qui fait converger les électrons sur l’anode centrale.

Les deux modes d’obtention des spectres de rayons X.
La méthode de détection en énergie (ED-XRF) comporte un détecteur qui peut discriminer chaque photon X en fonction de son énergie. La méthode séquentielle, quant à elle, permet d’obtenir un spectre en longueur d’onde (WD-XRF) suivant un montage goniométrique avec cristal et détecteur, mobile ou fixe (ex. : appareils multicanaux).

Spectromètre de fluorescence X à dispersion en énergie.
Agencement des différentes parties en prenant comme exemple le modèle MiniPal 4 de la société Philips analytical.

Quelques cristaux réflecteurs montés dans les goniomètres des spectromètres dispersifs en longueur d’onde et relation de Bragg.
Pour que les deux rayons 1 et 2 soient en phase, la différence de trajet optique doit être un multiple de λ. Quand cette condition est respectée entre les plans 1 et 2, elle l’est aussi pour tous les autres plans et l’effet global est donc renforcé. C’est le principe des interférences constructives. Chaque cristal permet d’explorer une plage de longueurs d’onde. Plus celle-ci est grande, plus le cristal choisi doit avoir une distance inter réticulaire importante, mais plus sa dispersion angulaire est petite.

Spectromètres séquentiels à cristal.
En haut, schéma réalisé d’après l’appareil SR300 de la société Siemens. Le collimateur primaire canalise le faisceau de rayons X, produits par une source puissante. Le collimateur secondaire sert à éliminer toute la lumière diffractée qui ne serait pas parallèle à la direction 2θ dans laquelle se trouve le détecteur. Ces composants sont constitués de feuillets métalliques parallèles. En bas, schéma inspiré du modèle ARL 9800, réunissant à la fois un montage goniométrique et des canaux fixes (couples cristal/détecteur) positionnés pour des éléments prédéfinis ; à droite spectre enregistré en fonction de l’angle 2θ obtenu avec ce type d’appareil.

Densimétrie X. % de transmission de deux films d’éléments légers.
Le film de 7μm de béryllium est souvent utilisé comme matériau pour confectionner les fenêtres des détecteurs en énergie. On remarque que pour une radiation de 1keV, (Na Kα par ex.) l’atténuation apportée par ce film est encore de 50 %.

Appareils de poing, type EDXRF.
a) Schéma explicatif du modèle Niton ThermoScientific. b) Spectromètre Tracer de la société Oxford instruments. c) Exemples de résultats fournis par ce type de spectromètre.

Spectre d’un échantillon de sol martien, obtenu par le Mars Rover Spirit en 2004 (documents NASA).






Expérience du « renversement des raies », de Kirchhoff.
Le schéma conventionnel du montage optique (collimateur, objectif) a été simplifié pour plus de clarté.

Quelques niveaux excités de l’atome de sodium.
Représentation simplifiée des niveaux d’énergie de l’atome de sodium. Origine des différentes radiations émises, compte tenu des règles de sélection. Valeurs indiquées en nm.



Exemples de courbes d’étalonnage en absorption atomique.
Droite d’étalonnage avec un appareil à effet Zeeman (voir § 8.2) pour le dosage du sodium à des concentrations sub ppb et courbe quadratique pour le dosage du zinc à des concentrations de l’ordre du ppm avec un appareil à brûleur. Cette dernière courbe montre que pour les concentrations de l’ordre du ppm la linéarité de l’absorbance n’est plus respectée. Les logiciels d’analyse quantitative en SAA proposent plusieurs types de courbes d’étalonnage.

Les diverses parties d’un appareil commercial d’absorption atomique monofaisceau.
Modèle IL 157, construit dans les années quatre-vingt. 1, source (lampe spectrale) ; 2, flamme du brûleur ; 3, monochromateur à réseau et 4, détecteur (photomultiplicateur). La source éclaire une fente située à l’entrée amont du système dispersif. La fente de sortie, en aval, est à proximité de la fenêtre du détecteur. Elle permet de sélectionner une étroite bande passante du spectre (Δλ de 0,2 à 1nm), qu’il ne faut confondre ni avec la largeur de cette fente de sortie, ni avec celle de l’image de la fente d’entrée.

Les deux types de sources en SAA.
La cathode est un cylindre creux dont l’axe de révolution correspond à l’axe optique de la lampe. À droite, représentation imagée de l’excitation des atomes de la cathode sous l’impact des ions néon. Lampe à décharge EDL. Les raies d’émission principales de l’arsenic. Ces montages ne sont pas adaptés à l’élément sodium (point de fusion trop bas) ou au mercure (état liquide), pour lesquels on utilise des lampes à vapeur métallique.

Comparaison des intensités transmises en SAA avec une source à continuum (1 et 2), et avec une lampe à raies spectrales (3 et 4).
Le rectangle figure l’intervalle de longueurs d’onde que “voit” le PM. Le signal de ce dernier est proportionnel à la surface des parties en blanc. Par cette astuce, « la résolution est dans la source », pour reprendre l’expression de Walsh, l’un des pionniers de l’absorption atomique.

Brûleur et système d’atomisation électrothermique.
a) Brûleur (d’après un document Perkin-Elmer) ; b)c)d) four graphite : tube graphite placé entre les deux électrodes, en accordement électrique transversal ; e) courbe de programmation de température avec profil absorbance-temps. Les deux premières étapes de cette programmation sont effectuées sous atmosphère inerte (balayage d’argon).

Dispositif hydrure.
Réservé à certains éléments, cet automate comporte un tube de mélange où l’hydrure du métal (ou du non-métal) est formé par action de l’hydrure de bore. Un courant d’argon extrait l’hydrure métallique formé (séparateur de gaz) pour l’entraîner dans un tube de silice porté entre 800 et 1000 degrés dans la flamme.

Modification de matrice.
Le nitrate d’ammonium ou l’acide EDTA augmentent la volatilité de certains éléments. Complexe volatil de type 1:1 entre une molécule d’EDTA et un ion Ni++.

Schéma d’un appareil mettant en jeu la correction par lampe au deutérium.
Le montage optique du type pseudo-double faisceau permet de s’affranchir de la dérive de la lampe pendant la période où elle n’est pas encore stabilisée. Le trajet optique de la lampe à deutérium se superpose par l’effet d’un miroir semi-transparent, à celui en provenance de la source à CC. La lumière qui atteint le détecteur provient en alternance soit de la voie de référence (b) soit de la voie échantillon (a). L’appareil mesure le rapport des intensités transmises par les deux faisceaux. Le domaine de correction est limité à la gamme spectrale de la lampe au deutérium, soit 180-420nm (d’après le schéma optique du modèle SpectraAA-10/20 de la société Varian).

Effet Zeeman simple.
Explication imagée du montage inverse/transverse constant à partir de l’absorption du cadmium à 228.8 nm. En 1, plan de polarisation perpendiculaire au champ magnétique, en 2, plan de polarisation parallèle au champ.

Correction à lampe pulsée.
Profils d’une raie d’émission en régime normal et régime forcé. Les rectangles noirs illustrent l’intervalle de longueur d’onde issue du monochromateur, impactant le détecteur pour ces deux situations. I0 correspond à la position sans échantillon sur le parcours lumineux. L’absorbance de l’analyte (Aana) est égale à l’absorbance totale (Tot.) minorée de l’absorbance du bruit de fond (BF).


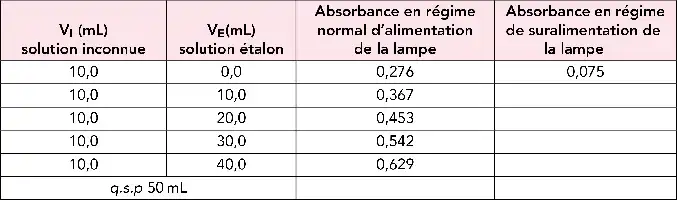


Transitions énergétiques pendant l’émission atomique.
Pour chaque atome il existe des centaines de possibilités de retour à l’état fondamental, chacune ayant une probabilité de se produire, si bien que pour le plus petit échantillon, qui comporte déjà un nombre énorme de ces atomes, on observe un spectre d’émission riche de centaines de transitions. À droite, exemple d’un spectre partiel d’une solution aqueuse contenant quelques ppm d’un sel de chrome.

Torche à plasma obtenu par couplage inductif.
Torche plasma présentée en position verticale (prise de lumière radiale) et en position horizontale (prise de lumière axiale). La torche est alimentée par un courant de radiofréquences (entre 27 et 50 MHz), à l’origine de la circulation des électrons dans le gaz inerte. Une torche consomme 10 à 15 L/min d’argon qui sert à la fois de gaz d’ionisation, de gaz de nébulisation et de gaz de refroidissement (une nécessité pour éviter que la torche ne fonde !). La température varie en effet de 9 000 à 2 000 K.

Nébuliseurs.
Modèles à flux croisés (1), concentriques (2) et parallèles (3). Contrairement aux deux autres modèles, ce dernier type n’est pas sujet au bouchage par d’éventuelles impuretés solides dans la solution. Celle-ci sort par un gros capillaire et s’écoule par gravité le long de la rainure en forme de V jusqu’à l’embouchure du gaz qui produit l’aérosol. En aval du dispositif nébuliseur on trouve une chambre de nébulisation comportant un drain, ce qui permet d’éliminer par gravité les plus grosses gouttelettes.

Dispositifs d’excitation par laser et par décharge luminescente.
a) Par laser (LIBS). Une impulsion laser de quelques nanosecondes génère un plasma dans lequel les atomes de l’échantillon bombardé sont ionisés. Après un court délai (ex. 500 ns), la lumière émise par le plasma refroidi est analysée par la partie spectrographe de l’appareil. b) Par décharge luminescente. L’échantillon, placé à la base d’une petite cavité cylindrique où règne une faible pression, est bombardé par des ions argon (générés par ionisation électronique). Les atomes détachés de la surface de l’échantillon sont ionisés ou excités par chocs des espèces neutres ou chargées présentes pour former un plasma « froid ». Le retour des atomes à leur état fondamental est à l’origine d’une émission de photons caractérisant l’échantillon (et l’argon). Ce dispositif est celui utilisé par la société Horiba.

Spectrométrie atomique ayant recours à un plasma induit par étincelage ou par laser.
Deux modèles de formation de plasma par ablation de surface. À gauche, poste mobile d’analyse industrielle par étincelage, généré au moyen du pistolet, relié par fibre optique au spectrophotomètre situé dans la console (modèle ARC Met-8000 de la société Metorex), À droite, spectromètre de terrain ayant recours à un laser pulsé (2,4 GHz) focalisé sur une surface microscopique de l’échantillon (technique LIBS pour Laser Induced Breakdown Spectroscopy). Modèle m-Pulse reproduit par autorisation de la société Oxford instruments.

Les trois causes fondamentales d’élargissement des raies atomiques.
Représentation d’une transition entre un niveau fondamental stable, dont la durée de vie est infinie et un niveau excité de très courte durée de vie (Heisenberg) ; effet de la vitesse (Doppler) ; effet de la pression (Lorentz).

Agencement de l’optique d’un appareil à réseau concave et polychromateur à grande distance focale.
Lorsque les raies à détecter sont trop proches on peut avoir recours à des miroirs de renvoi. Plusieurs polychromateurs sont quelquefois associés. En haut, à droite, principe du cercle de Rowland. La partie optique de ces appareils doit être montée sur un socle métallique très stable.

Principe de la dispersion dans le plan focal d’un montage associant un réseau échelle et un prisme.
Pour plus de clarté, l’optique associée (lentilles collimatrices et focalisatrices) n’a pas été représentée. Comparaison d’un réseau de diffraction échelle et d’un réseau conventionnel. L’angle de plus forte luminosité du réseau est repéré par l’angle de « blaze » θ (ce terme pouvant se traduire par miroiter). La formule de base indique quelles sont les longueurs d’onde renvoyées par le réseau pour un angle d’incidence α par rapport à la normale du réseau et pour une direction d’observation faisant l’angle β (le signe +si l’observation est du même côté que le rayon incident par rapport à la normale, et – dans le cas contraire). La résolution peut atteindre celle d’un appareil qui étalerait le spectre sur une longueur de 2m dans le plan focal.

Schéma optique d’un spectromètre multicanal à réseau de diffraction échelle.
Pour plus de clarté, seule la partie centrale du faisceau issu de la source 1 est représentée (ce faisceau devrait recouvrir tout le miroir 2). Le réseau échelle 5 sépare les radiations présentes dans la source dans le sens horizontal (en x). Le prisme dévie ensuite ces radiations dans le sens vertical (en y). Le parcours de trois lignes spectrales différentes est schématisé. Les images du trou d’entrée 2 sont dans le plan focal 8. Pour détecter ces radiations on peut installer soit des photomultiplicateurs de taille réduite en des endroits spécifiques, soit un capteur à matrice de 105 à 106 photodiodes (CID ou CCD) pour couvrir toute l’étendue spectrale de 190 à 800 nm. Une étude complète de l’échantillon est alors possible (dessin exécuté d’après un schéma du modèle 7000 PU de la société Unicam.). En haut à droite, image de la répartition des spots lumineux générés par un échantillon sur une matrice de capteurs.

Dispersion linéaire et dispersion linéaire réciproque (dispersion inverse).
Un spectromètre d’émission atomique comporte un ou plusieurs réseaux optiques gravés qui dispersent les différentes radiations dans un plan focal. La dispersion réciproque est accessible expérimentalement.

Analyse d’un échantillon de sol martien par émission atomique après excitation par laser (technique LIBS).
Un des nombreux spectres du sol martien réalisés à partir des données transmises par le Chem-Cam, module embarqué sur le véhicule Curiosity (2012). Les pics principaux entre 240 et 340 nm sont identifiés.

Photométrie de flamme.
Schéma de principe d’un photomètre de flamme. Modèle PFP7 de la société Janway qui permet de doser 5 éléments prédéfinis. Une liste d’éléments quantifiables par photométrie de flamme (le calcium est dosé par son émission à 622 nm, due à l’oxyde CaO). La gamme dynamique de ces dosages assez faible (2 décades).



Présentation conventionnelle d’un spectre de RMN du proton (1H) d’un composé organique.
Ici, spectre de la butanone [CH3(C = O)CH2CH3] avec, en superposition, la courbe d’intégration, qui permet d’évaluer les aires relatives des principaux groupes de signaux repérés sur le spectre. La nature de l’échelle des abscisses sera expliquée plus loin.

Effet d’un champ magnétique sur un noyau de nombre de spin 1/2 présent dans un composé en solution.
Si le noyau est dans la partie supérieure de l’échantillon, non soumise au champ magnétique extérieur, \begin{equation}\vec{\mu}\end{equation} n’a aucune orientation préférentielle. En revanche, dans la partie centrale baignant dans le champ, \begin{equation}\vec{\mu}\end{equation} balaie la surface d’un cône de révolution dont l’axe est aligné avec \begin{equation}\overrightarrow{B_0}\end{equation}. La projection de \begin{equation}\vec{\mu}\end{equation} est dans le même sens que \begin{equation}\overrightarrow{B_0}\end{equation} ou dans le sens opposé.


Représentation de l’éclatement entre les niveaux énergétiques d’un noyau de nombre de spin I = 1/2 placé dans un champ magnétique.
Les quatre valeurs choisies du champ B0 correspondent, pour le proton, à des appareils commerciaux dits à 60, 200, 300 et 400 MHz. (B0 représente la densité de flux magnétique exprimée en Tesla : 1T équivaut à 10 000 gauss).

Précession et magnétisation.
a) Instantané figurant le mouvement de précession du vecteur spin de 5 noyaux indépendants dans le champ magnétique extérieur ; b) Vecteur magnétisation macroscopique résultant des orientations individuelles d’un grand nombre de noyaux. L’échantillon en solution devient donc faiblement aimanté. \begin{equation}\overrightarrow{M_0}\end{equation} est un objet auquel s’appliquent les lois de l’électromagnétisme.


Spectre de RMN d’un échantillon d’eau placé dans un récipient de verre borosilicaté, observé à une fréquence de 5 MHz (le champ est exprimé en Tesla ; document Varian).
Il n’existe pas d’appareils commerciaux de ce type, la RMN étant une méthode réservée à des examens plus approfondis que rechercher la composition élémentaire des d’échantillons.

Représentation d’une onde électromagnétique et effet sur un noyau.
En tout point du plan xOy la composante magnétique de l’onde peut être dissociée en deux vecteurs de normes moitié \begin{equation}\overrightarrow{b_1}\end{equation} et \begin{equation}\overrightarrow{b_2}\end{equation} tournant en phase à des vitesses opposées. Ici, seul \begin{equation}\overrightarrow{b_2}\end{equation} peut basculer le noyau dans la population E2.

Basculement du vecteur magnétisation → M0 et retour à l’équilibre après résonance.
Représentation imagée où ne sont figurés que les vecteurs individuels marquant le déséquilibre des populations (numériquement). Un observateur tournant à la fréquence de précession verrait le vecteur magnétisation basculer d’un angle a. Retour progressif de la projection → Mz de → M0 à la situation d’origine.

Signal FID de la fluoroacétone (13C) obtenu avec un appareil à onde pulsée.
Le signal I = f(t) est converti en spectre classique I = f(ν) par transformée de Fourier. L’in-terférogramme, capté en quelques secondes d’enregistrement, peut être accumulé plusieurs dizaines de fois avant d’être transformé. Cette accumulation de FID permet d’améliorer le rapport signal/bruit de fond.

Appareil de RMN à onde continue.
Noter l’agencement des différents enroulements au niveau de la sonde entre les pièces polaires de l’aimant et les orientations des différents champs. À droite un enregistrement historique (1951) du spectre des protons de l’alcool éthylique. Les trois signaux correspondent aux atomes d’hydrogène du CH3, du CH2 et du OH. On comprendra mieux après avoir lu le §8.

Les deux processus de relaxation des noyaux.
Évolution au cours du temps des paramètres de relaxation spin/spin et spin/réseau.

Déplacements chimiques de quelques composés en RMN du proton. Effets d’écran avec un appareil qui fonctionne à une fréquence fixe.


Effets de résonance pour les composés carbonylés en RMN 13C.
Si on compare le carbonyle d’une cétone avec celui d’un ester, on note que pour ce dernier le carbone est moins déblindé que celui de la cétone dont le caractère est plus électropositif. En RMN du 13C le signal du carbonyle se situe vers 205 ppm pour une cétone et pour un ester, vers 165 ppm.

Effets d’anisotropie et de champs locaux induits.
La présence de liaisons π, est traduite ici sous forme de zones où on a un effet de blindage (+) ou de déblindage (–). Les protons éthyléniques sont à l’extérieur d’une sorte de double cône de protection et les protons aromatiques subissent l’effet de la circulation des électrons de cycle dans deux volumes toriques.


Diagramme de couplage de la molécule HF en RMN du proton.
Situation de principe dans l’hypothèse où il n’y a pas de couplage avec l’atome de fluor et situation réelle. Les valeurs ν1 et ν2 diffèrent de la valeur de J (Hz).

Représentation des différents états de spin des 5 protons d’un groupement éthyle.
Sur une même rangée se trouvent réunis les états de spin produisant le même effet sur les noyaux voisins. L’échantillon comportant un très grand nombre de molécules identiques, celles-ci se répartissent en plusieurs populations donnant chacune un signal pondéré comme le nombre d’états par rangée de ce schéma.

Triangle de Pascal et son application à la RMN pour \begin{equation}I=\frac{1}{2}\end{equation}

Spectres des 4 protons aromatiques de l’aspirine.
La figure reproduit le spectre d’un échantillon d’aspirine, obtenu avec deux appareils, l’un fonctionnant à 90 (solvant CDCl3) et l’autre à 400 MHz (solvant C3D6O). La sensibilité de la RMN croît comme B03/2.

Caractéristiques définissant un système AB.
a-d) Modification graduelle d’un système de deux protons couplés, à mesure que la valeur du rapport Δ(ν/J diminue. Aspect typique et formules utilisées pour l’analyse d’un système AB.

Nomenclature des spectres.
Le spectre 1H de la 3-chloro 4-méthylpropiophénone peut être vu comme résultant de la superposition des signaux de plusieurs sous-ensembles indépendants, facilement reconnaissables. Ainsi le groupement éthyle (comme l’éthoxyle) constitue un système A2X3 ; les protons de l’éthanal forment un système AX3 tandis qu’un groupe vinyle sera un système ABC, AMX ou ABX suivant l’exemple étudié.

Expérience de découplage de spin sur la butanone.
Modification du spectre du proton (1H) par : a) irradiation du CH2 à 2,47 ppm; b) irradiation du CH3 (de l’éthyle) à 1,07 ppm. Comparer avec l’enregistrement de la butanone (fig. 15.1). Pour un composé simple tel que celui-ci, ce type d’expérience n’a qu’un intérêt illustratif. Par contre une expérience de double résonance permettrait de déterminer avec précision les différents couplages de l’aspirine (fig. 15.17).

Séquences DEPT sur le 2-carène.
Cet exemple illustre tout l’intérêt de ces séquences quand il s’agit de déterminer les structures de composés a priori inconnues.

Spectre 13C du CDCl3.
Le deutérium, pour lequel I = 1, conduit à 3 valeurs de m (−1, 0, 1) et par suite à trois pics d’égale intensité puisqu’il n’y a qu’un seul atome de deutérium.

Spectre COSY de l’heptanone-3.
Sur cette représentation on voit sur une même horizontale les protons qui donnent lieu à des couplages entre eux. Ainsi, on voit apparaître le couplage 3J entre les protons portés par le carbone 6 et les protons portés par les carbones 5 et 7. On pourra retrouver sur différentes horizontales les autres couplages existants.

Spectre NOESY de l’éthylbenzène.
Le résultat d’une expérience NOESY sur cette molécule fournit un exemple simple qui permet de remarquer qu’en plus de la corrélation COSY entre les atomes d’hydrogène situés sur les carbones 1 et 2, il apparaît une tache de corrélation plus lointaine avec certains des hydrogènes situés sur le cycle aromatique.

Spectre du 13C de l’éthylbenzène découplé des protons et spectre HSQC.
Chaque atome de carbone donne un seul signal, chaque carbone étant alors représenté par un singulet. Ces spectres découplés « large bande », plus simples, donnent moins d’informations. Sur cette figure a été reproduit également le résultat d’une expérience 2D de type HSQC de cette même molécule. On notera que le carbone 3 ne portant pas d’atome d’hydrogène, ne peut donner de tache de corrélation.

Spectres de RMN de la monofluoroacétone.
Un exemple de couplage hétéronucléaire. L’unique atome de fluor de cette molécule conduit à un triplet avec le CH2 et à un quadruplet avec le méthyle. Le signal résultant est donc un triplet de quadruplets (l’échelle des déplacements chimiques est positionnée par rapport à FSiCl3).

Positions de quelques signaux en RMN du fluor et du phosphore.
La figure reproduit le spectre d’un échantillon d’aspirine, obtenu avec deux appareils, l’un fonctionnant à 90 (solvant CDCfi) et l’autre à 400 MHz (solvant C3D6O). La sensibilité de la RMN croît comme Bo3/2.

Enregistrement obtenu au cours d’une expérience CLHP couplée à une détection par RMN 1H.
Séparation de deux dipeptides (5 μg chacun) avec identification des signaux. Pour plus de clarté, les pics des solvants vers 2 et 5 ppm (acétonitrile et eau) ont été éliminés (selon Sweeder et al, Analyt. Chem., 1999, 71(23), 5335).

Spectre de RMN d’un mélange d’acétone A et de benzène B.
La figure reproduit le spectre d’un échantillon d’aspirine, obtenu avec deux appareils, l’un fonctionnant à 90 (solvant CDCfi) et l’autre à 400 MHz (solvant C3D6O). La sensibilité de la RMN croît comme Bo3/2.

Spectre d’un échantillon auquel on a ajouté un composé de référence R.
Le signal SX appartient au composé à doser et le signal SR, au standard interne.

Détermination de l’indice de graisse solide par décroissance du FID.
La décroissance du signal après l’impulsion de radiofréquence permet ici de déterminer le pourcentage de graisse solide dans un échantillon sans préparation. Un facteur correctif intervient pour conduire à la valeur du signal sitôt après la fin de l’impulsion. Au centre, un échantillon de graines placées dans un tube de verre prêt à être introduit dans l’appareil (mesure du taux d’humidité). À droite, appareil de faible encombrement comportant un aimant permanent de 1 T utilisé pour quantifier l’eau et les matières grasses dans les échantillons (modèle QMC de la Société Oxford Instr.).

L’intérieur d’un kiwi.
L’une de ces images est une photo d’un kiwi (coupé en deux) et l’autre une image obtenue (sans le couper), par IRM du proton d’un autre kiwi. Le signal de RMN varie selon les caractéristiques de T1, de T2 et de la densité des protons en chaque point (la photo est à droite).

Déplacement chimique des principaux types de protons des molécules organiques en RMN. (reproduit avec l’autorisation de la société Spectrométrie Spin et techniques)




Spectre defragmentation et spectre de masse présentés sous forme graphique ou tabulaire.
a) Spectre de fragmentation du méthanol ; b) représentation non conventionnelle du même spectre sous forme d’un diagramme circulaire : statistiquement, pour 321 ions formés, il y en a 100 de masse 31 u, 72 de masse 29, etc. Les divers ions constituent autant de populations différentes ; c) partie d’un enregistrement haute résolution d’un composé M présentant deux ions de masse voisine (l’un par perte de CO et l’autre de C2H4).


Pouvoir de résolution.
À gauche, situation de principe pour définir ce paramètre dans le cas d’un pic isolé. Suivant les constructeurs, la largeur du pic est mesurée soit à 50% soit à 5% de sa hauteur. À droite, exemple de spectre basse résolution d’un échantillon de plomb. La valeur trouvée pour le pouvoir de résolution dépend beaucoup du composé et de la masse choisis.

Pouvoir de résolution établi sur des spectres expérimentaux.
À gauche exemple d’un enregistrement correspondant à un très grand pouvoir de résolution (appareil à résonance cyclotronique). En haut, à droite, le signal brut d’un appareil à filtre quadripolaire (pouvoir de résolution, 450). À droite présentation conventionnelle de ce même enregistrement (résolution, 1 u).


Installation du système de pompage d’un spectromètre de masse.
La pompe turbomoléculaire est directement fixée à l’analyseur du spectromètre par sa large ouverture, alors que la pompe de prévidage est généralement à quelque distance pour éliminer de possibles vibrations.

Pompe turbomoléculaire.
Schéma de principe. L’effet de pompage est obtenu par une alternance de rotors tournant à grande vitesse et de stators (fixes). L’illustration de droite montre l’empilement des rotors/stators d’une pompe de ce type (extrait d’une documentation de la Société Edwards).

Ionisation électronique.
Impact d’un électron sur une molécule m, avec apparition de l’ion parent et d’ions secondaires \begin{equation}\mathrm{m}_1^{+}\end{equation} et \begin{equation}\mathrm{m}_2^{+}\end{equation} en filiation directe. Les fragments neutres, \begin{equation}\mathrm{m}_1^{\prime}\end{equation} et \begin{equation}\mathrm{m}_2^{\prime}\end{equation}, ne sont pas détectés. Illustration dans le cas du benzène. Schéma d’une chambre d’ionisation (ou source d’ions). En superposant un champ magnétique dirigé parallèlement aux électrons, ces derniers suivent un mouvement en spirale, qui améliore l’efficacité de ce canon à électrons.

Influence de l’énergie des électrons sur la fragmentation.
Exemple de l’acide benzoïque. Courbe de l’efficacité de l’ionisation en fonction de l’énergie des électrons. On remarquera que les spectres sont normalisés c’est-à-dire que le pic le plus intense prend la valeur 100. Certains disent que c’est une application de la méthode de Procruste (selon la légende grecque, le brigand Procruste forçait les voyageurs à s’allonger sur un lit et selon leur taille il leur coupait les pieds ou au contraire tirait sur leurs membres pour les mettre aux dimensions du lit).

Ionisation chimique.
Formation à partir du méthane d’espèces cationiques et réaction sur les molécules M du composé. La dernière équation correspond à l’action du cation tert-butyle, issu de l’isobutane par perte du proton tertiaire. Le symbole +· signifie qu’il s’agit à la fois d’un radical (nombre impair d’électrons) et d’un cation.

Techniques FAB et MALDI.
a) Principe de formation d’un atome rapide et neutre de xénon par collision ; b) formation d’atomes rapides d’argon dans une chambre de collision et bombardement de l’échantillon (canon FAB) ; c) ionisation par laser (Matrix Assisted Laser Desorption Ionisation). L’impact du photon est comparable à celui d’un atome lourd. Par un mécanisme mal connu il se produit une désorption et photo-ionisation des molécules.

Ionisation à pression atmosphérique par electrospray (« ionspray »).
Le capillaire de sortie porté à un potentiel élevé, conduit à un brouillard chargé (1). Les gouttelettes, en s’évaporant, provoquent une augmentation de la densité de charges électriques (2) au point qu’elles explosent en expulsant des molécules de l’analyte porteuses de plusieurs charges (3). Le diazote améliore le processus de concentration (4).

Ionisation, à pression atmosphérique par ionisation chimique.
(1) L’échantillon, sous forme de spray, se mélange aux ions issus d’un gaz réactant tel le diazote ionisé par une aiguille portée à un potentiel très élevé (effet corona). (2) Les molécules ionisées de diazote ou de solvant captent à leur tour des molécules d’analyte pour conduire à des amas (« clusters ») (3). Ceux-ci sont détruits par un courant de diazote qui permet un transfert des charges sur l’analyte.

Ions moléculaires multichargés.
Spectre obtenu à partir du cytochrome C (cœur de cheval), protéine de 12 360 Da, par la méthode électrospray. Entre 2 pics consécutifs, la charge de l’ion varie d’une unité. À droite, amas isotopique du cytochrome en haute résolution. On peut retrouver, à partir de ces spectres et par deux méthodes indépendantes, des valeurs très proches de la masse moléculaire ainsi que le nombre de charges portées par les ions (cf. § 8.3).


Spectrographe de Bainbridge à déflexion magnétique de 180 °, comportant un filtre de vitesse.
Ce dernier permet de s’affranchir de la quasi-impossibilité d’avoir des faisceaux hétérogènes monocinétiques. Dessin d’un enregistrement sur plaque photographique du spectre du néon. Les deux séries de taches résultent de l’arrachement de un ou de deux électrons aux différents isotopes de l’élément néon (m/z : 20Ne+, 21Ne+·, 22Ne+· ; 20Ne++, 21Ne++, 22Ne++) Mais la paternité de ces montages revient à J.-J. Thomson qui, dès 1913, prouve l’existence des isotopes du néon ainsi qu’à Aston, pour le soufre et le chlore.

Un appareil conçu autour d’un analyseur électromagnétique de type BE.
Modèle JMS 700 de la Société Jeol. On reconnaît sur cette photographie, la forme caractéristique de l’électroaimant (secteur magnétique). Le détecteur est à l’extrême droite et la source de l’appareil est à gauche de la photographie. Dans la configuration représentée, on notera également que l’appareil est en aval d’une installation de CPG (reproduit avec l’autorisation de la société Jeol, Japon).

Effet de la tension accélératrice sur la dispersion des vitesses des ions d’une même masse.
En admettant que la dispersion des énergies avant et après accélération est la même (ΔE1 = ΔE0), la plage de vitesse après accélération Δν1 est beaucoup plus petite. (ΔE1 = ΔE0 mais Δν1 ≪ Δν0).

Principe de l’agencement d’un spectromètre de masse EB à double focalisation.
R′ et R mesurent quelques décimètres. Les lois de la physique et les relations rappelées dans ce chapitre, utiles pour concevoir ces appareils, ne servent pas au calcul des masses décelées par l’appareil. Tous les SM adoptent le principe de mesures comparatives. Chacun d’eux doit être étalonné avec des masses connues, comme celles des ions de fragmentation de la perfluorotributylamine.

Spectromètre à temps de vol à parcours direct et principe du réflecteur d’ions.
1) Échantillon et porte-échantillon ; 2) dispositif MALDI d’ionisation ; 3 et 3’) grilles d’extraction et d’accélération (ddp 5000 V) ; 4) grille de contrôle ; 5) collecteur plan à microcanaux ; 6) sortie du signal. Le dessin du bas représente un réflectron dont le « miroir électrostatique » égalise les temps de vols (quelques μs) des ions de même masse mais dont les énergies initiales diffèrent. Les largeurs de pics sont de l’ordre de 10−9 s et la résolution atteint désormais 15 à 20 000.

Représentation d’un quadripôle linéaire.
Noter le raccordement des barres 2 à 2. Ces barres de petite taille (environ celle d’un crayon) demandent un usinage particulier pour les rendre hyperboliques dans la partie centrale au filtre. À droite, figuration du champ dans l’espace quadripolaire à partir du tracé d’une série d’hyperboles équipotentielles. Les barres qui sont alternativement positives et négatives entraînent les ions dans un mouvement en spirale.

Filtre quadripolaire.
Suivant leur masse, les ions répondent plus ou moins facilement aux sollicitations du champ variable. Leur amplitude en xy ne doit pas dépasser r0. Les ions entrent dans le filtre avec une énergie cinétique de quelques dizaines d’eV seulement. Exemple d’une ligne de balayage pour un rapport U/V fixe.

Exemple de gaz résiduels dans un vide poussé.
L’enregistrement est en mode Δm constant sur toute l’étendue des masses. Sonde quadripolaire dont la source est directement immergée dans l’enceinte à vide (d’après une documentation de la société Inficon, EUA).

Spectromètres à piégeage d’ions.
a) Agencement des électrodes d’une trappe à ions ; b) dessin en perspective des électrodes terminales et de l’électrode annulaire centrale.


Le principe du spectromètre de masse à résonance cyclotronique.
a) Trajectoire de base suivie par un ion soumis à une induction magnétique \begin{equation}\overrightarrow{B}\end{equation} compte tenu des orientations choisies. b) aspect simplifié d’une cellule de piégeage des ions (les plaques 5 et 6 sont excitatrices, les plaques 3 et 4 piègent les ions et les plaques 1 et 2 sont détectrices). Les ions sont formés soit dans la cellule soit en amont de celle-ci. c) principe de la détection : 1, trois ions en attente ; 2, impulsion de fréquence ν1 : seul l’ion m1 est excité ; 3, après excitation, l’ion m1 tourne sur une orbite plus grande ; 4, pour un échantillon réel contenant un grand nombre d’ions m1, ceux-ci se mettent à tourner en phase. Ils produisent, sur les plaques détectrices 1 et 2, l’effet d’un vecteur champ électrique tournant à la même fréquence ν1 dont l’intensité est fonction de leur concentration. Si les trois ions m1, m2 et m3 ont été simultanément excités, le signal en sortie sera un interférogramme. Un calcul de Fourier conduit au spectre habituel.

Principe d’un appareil SM/SM à 3 quadripôles en série.
Dans ce montage appelé QQQ, le quadripôle central a un simple rôle focalisateur. Situé dans la chambre de collision, il est alimenté par une tension alternative uniquement (absence de tension continue). La pression à cet endroit de l’appareil est plus élevée. Avec ces instruments on peut étudier des mélanges comme on le ferait avec la technique CPG/SM. Les applications sont devenues très nombreuses sachant que le coût de ces appareils a beaucoup diminué.

Détecteurs en SM.
a) Modèle à dynodes séparées à film actif (reproduit avec l’autorisation de la société ETP Scientific Inc.) ; b) modèle à dynode continue. Représentation d’un channeltron : la forme en entonnoir de la cathode permet la récupération des ions issus de trajectoires légèrement différentes. La courbure a pour effet d’éviter que des ions positifs formés par impact des électrons sur des molécules résiduelles, ne prennent trop d’accélération avant de frapper à nouveau la cathode et ne génèrent ainsi d’autres électrons (feed-back du channeltron) ; c) détail de la cathode de conversion. Multiplication des électrons dans un microcanal ; d) un multiplicateur à galette. Système de collecteur multicanaux (microchanneltron) qui permet de localiser l’impact des ions (d’après illustrations de Galileo USA).

Masses atomiques des isotopes des principaux éléments rencontrés en chimie organique.
La masse nominale d’un élément est la masse entière de son isotope naturel et stable le plus abondant. La masse nominale d’un ion est la somme des masses nominales des atomes présents dans sa formule empirique (ex. HCl+ = 36 u).

Spectre profil d’un anticorps de masse moléculaire élevée.
Ce spectre a été obtenu par électrospray. Sans utiliser les valeurs des charges reportées audessus de chaque signal, le calcul décrit dans le texte conduit à partir des seules valeurs de M1 = 2 680 et de M2 = 2 729 à M = 150 080 Da.

Spectre de fragmentation de la butanone obtenu par ionisation électronique.
Schéma expliquant la formation des principaux ions fragments. L’ion moléculaire conduit à l’ion acétyle CH3CO⌉+ (m/z = 43), dix fois plus intense que l’ion 57, CH3CH2CO⌉+. Les demi-flèches symbolisent le transfert d’un seul électron.



Pics métastables. Aspect théorique des trois pics constituant une transition métastable.

Apparition d’ions métastables au cours de la fragmentation de la théobromine.
L’ion moléculaire (180 u), par perte de CONH• (43 u) conduit à l’ion de masse 137 u, transition métastable, accompagnée du pic diffus à 104,3. De même la perte de CO (28) à partir de l’ion 137, est un second exemple de transition métastable qui apparaît à m/z = 86,7.

Coupure de la liaison peptidique.
Sur cet exemple d’un tétrapeptide traité par ionisation de type ESI, la protonation peut se fixer sur un atome d’oxygène ou d’azote entraînant la formation de très nombreux fragments. Les lettres a, b, c, x, y, z font référence à la nomenclature des fragments issus d’un même mécanisme en se déplaçant le long de la chaîne polypeptidique (sur la figure ce serait a3, b3, c3, x2, y2, z2), a, b, c pour les ions N terminaux et x, y, z pour les ions C terminaux. Le grand nombre de fragments rend leur identification difficile. Des logiciels viennent au secours des utilisateurs.

Ionisation par torche plasma et enregistrement obtenu par la méthode ICP/SM.
Environ 10% des ions générés dans le plasma passent dans le spectromètre de masse. Spectre d’un mélange de quelques éléments métalliques enregistré entre les masses 50 et 68 u. Noter la présence des différents isotopes des éléments concernés. La résolution est ici insuffisante pour distinguer les isobares, c’est-à-dire des ions provenant d’éléments différents mais ayant pratiquement même masse et qui se superposent donc sur le graphe. Un espace sous forme de chambre de collision est quelque fois intercalé entre la chambre d’introduction et le quadripôle pour décomposer les interférences polyatomiques (ArO, Ar2).







Dosage par CLHP/SM de la caféine par dilution isotopique.
L’isotope stable utilisé comme traceur est le deutérium D. La caféine-d3 est issue de la méthylation de la 1,3-diméthylxanthine par ICD3.


Les différentes étapes d’un test immunoenzymatique de type ELISA avec compétition (voir texte).
En analyse clinique, il existe de nombreux dosages de ce type.

Relation concentration/absorbance des tests ELISA du type décrit.
Le rapport des deux espèces en solution ou fixées sur la paroi est le même (ici, il vaut 3). La linéarité en échelle semi-logarithmique n’est atteinte que dans un domaine étroit de concentrations.

Technique EMIT.
Les enzymes sont souvent utilisées comme marqueurs car elles se comportent comme des catalyseurs qui amplifient la production d’espèces détectables.

Schématique de l’activation neutronique.
Quand un neutron agit sur un noyau cible, il se forme un isotope qui peut être instable. Dans ce cas, l’isotope formé émet instantanément un rayonnement γ et passe ainsi dans une configuration plus stable. Puis cet isotope radioactif décroît par émission d’un électron et d’un nouveau rayonnement γ caractéristique ; illustration avec comme cible, un atome d’argent 109.

Une application non conventionnelle de l’activation neutronique.
Lorsqu’il s’agit de détruire des obus d’arme chimique, il arrive qu’on utilise l’activation neutronique pour en connaître le contenu. Ce dessin, reproduit avec l’autorisation de la société EG & G Ortec, illustre le principe général de la méthode. En cartouche, une petite partie du spectre γ d’un obus contenant un agent chimique, obtenu en quelques minutes.

Trois molécules marquées en un seul site avec le 14C.
Ce radio-isotope a une période suffisamment longue qui permet un stockage aisé et évite de prendre en compte la diminution d’activité au cours du dosage par décroissance naturelle.
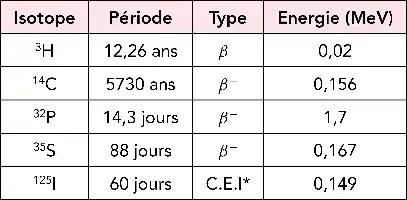

Trois molécules marquées en un seul site avec le 14C.
Spectres d’émission de fluorescence obtenus par excitation.

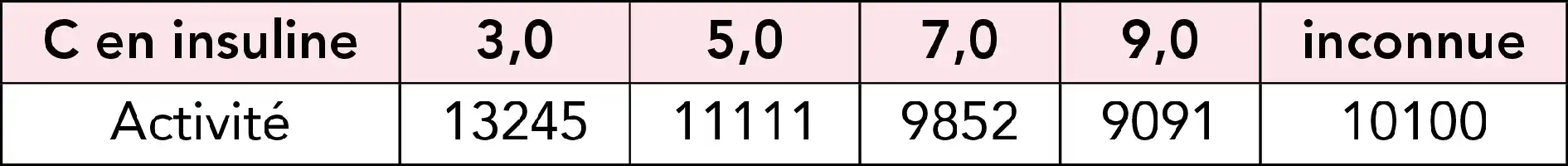


Les méthodes de microanalyse de Pregl et de Simon.
Des générations d’étudiants se souviennent avoir déterminé, avec quelle appréhension, des formules de composition à partir des masses de CO2 et d’eau piégés dans les absorbeurs démontables. Dans la version plus récente de Simon, les gaz de réaction passent sur de la poudre de cuivre pour réduire les oxydes d’azote en diazote. Trois détecteurs à conductibilité thermique (TCD) permettent d’avoir trois signaux témoignant de H2O, CO2 et N2. Le dosage des trois éléments demande environ 10 minutes.

Appareil de microanalyse avec détection chromatographique.
La colonne à remplissage sépare les 4 constituants gazeux entraînés par un courant d’hélium utilisé comme gaz vecteur. Par un étalonnage préalable, on peut déduire de l’aire des pics la concentration de chacun de ces 4 éléments dans l’échantillon.

Appareil dédié aux analyses d’azote.
Cet appareil est une adaptation moderne de la méthode de Dumas révisée par le four vertical et la détection au moyen d’un catharomètre (gaz vecteur He).

Analyseur d’azote.
Modèle ND20 reproduit avec l’autorisation de la société Skalar. Ce type d’appareil est utilisé pour le dosage des protéines. Détection par chimifluorescence.

Analyseurs de mercure par AAS ou FAS à vapeur froide.
a) Appareil par absorption atomique ; b) par fluorescence atomique.

Spectromètre de mobilité ionique (IMS).
Les ions sont admis de manière répétitive dans le tube analyseur en contrôlant la polarité de la grille amont. Exemple d’enregistrement obtenu à partir de deux explosifs, montrant ainsi que l’on peut attribuer des profils différents à chacun d’eux. Modèle de terrain Bruker RAID-100. Agencement d’une installation réunissant un IMS et un TOF.


Chaîne électrochimique de mesure avec une électrode ionique sélective (EIS).
La membrane perméable à l’ion choisi crée un potentiel variable suivant la concentration de l’analyte (ionique) en solution. Les autres potentiels sont imposés par construction. La force ionique de l’électrolyte support (qui ne doit pas réagir sur l’analyte !) doit être beaucoup plus grande que celle de l’échantillon contenant l’analyte. Les mesures se font à l’aide d’ionomètres. Le § 4 donne des indications sur la pratique de ce type de dosage.

Électrode combinée de verre pour la mesure du pH.
La concentration en ions H+ est accessible à partir de la différence de potentiel qui apparaît entre une électrode de verre et une électrode de référence (ici une électrode Ag/AgCl). Détail de la membrane, vue en coupe, perméable aux ions H+. Quand un ion H+ forme une liaison silanol, un ion sodium part en solution pour conserver l’électroneutralité de la membrane. Une électrode combinée de présentation classique, l’électrode de référence entourant l’électrode de verre, sauf à son extrémité. La jonction permet la migration des ions sans que les liquides de part et d’autre ne se mélangent.

Cellule électrochimique et EIS.
Cellule de mesure composée d’une EIS pour l’ion fluorure avec une électrode de référence à double jonction. Des 2 côtés du monocristal de LaF3, élément sensible de l’électrode, naît 1 équilibre LaF3 ⇌ LaF2+ + F−. L’une des faces devient positive par rapport à l’autre. Pour éviter l’osmose de KCl dans la solution à doser, l’électrode de référence est entourée d’une seconde chambre qui contient un électrolyte auxiliaire non interférant. Ainsi KNO3 1M est très utilisé pour F−, Cl−, I−, CN−, S− ou Ag+.). Les réalisations, quelquefois très compactes ne sont pas toujours faciles à visualiser. À droite EIS “tout solide” : pour l’ion chlorure.

Électrode à membrane liquide pour l’ion Ca2+ et composés organiques à usage d’ionophores.
Deux ionophores commerciaux chélatants : le « dioctyl » phosphate, sélectif de l’ion Ca2+et l’ionophore K2 sélectif de l’ion K+. Cette dernière molécule est représentative, par sa sous-structure d’éther-couronne, d’autres composés macrocycliques utilisés à cette fin.

Capteur potentiométrique de gaz dissous.
Schémas avec deux électrodes séparées ou une seule électrode combinée, réunion d’une EIS et d’une ERE (ici une électrode de pH). L’ensemble constitue une cellule électrochimique pour la mesure du CO2 dissous. L’espèce mesurée est H+, tous les acides ou bases sont donc des interférants éventuels. La sélectivité repose sur le choix de la membrane.

Valeurs de pentes idéales (298 K) pour quelques ions.
*S pour Slope (pente, en anglais)

Exemple de dosage par potentiométrie directe.
La courbe d’étalonnage de l’électrode spécifique de l’ion chlorure a une pente proche de la pente théorique. La gamme de mesure des différentes électrodes spécifiques s’étend sur 4 à 6 décades suivant les ions. Le cartouche représente les facteurs de pentes théoriques selon la valence de l’ion.



Montage de principe d’une cellule voltampérométrique.
a) montage à tension continue dans lequel aucune intensité ne passe par l’électrode de référence (par suite de son impédance élevée) ; b) un modèle d’électrode indicatrice (dite électrode de travail à goutte de mercure). Beaucoup de métaux peuvent être réduits à la surface du mercure avant que H+ ne soit réduit à son tour ; c) schéma de raccordement avec un potentiostat. Ce montage électronique permet de maintenir le potentiel d’une électrode de travail à un niveau constant par rapport à une électrode de référence ; d) domaines d’utilisation des quatre principales électrodes de travail (CV pour abréviation de carbone vitreux). La plage d’utilisation du mercure s’étend encore plus loin du côté cathodique dans un électrolyte comme KCl ou NaOH (- 2 V). Bien qu’il y ait recouvrement des plages d’utilisation entre les électrodes, leurs sensibilités peuvent être très différentes.

Évolution du diamètre de la goutte de mercure en fonction du temps. Mode DME à goutte renouvelée, SDME à goutte stabilisée et HDME à goutte pendante.

La vague polarographique.
Polarogramme d’une solution à 10 ppm de Pb++ dans KNO3 0,1M, obtenu avec une électrode à goutte de mercure croissante. La position médiane de la vague (ici -0,35 V) est caractéristique du plomb et la hauteur du palier, de sa concentration. Pour un meilleur rendu de la courbe, les oscillations sont ici amorties. À droite, construction graphique pour la mesure de iD. Seule l’enveloppe du polarogramme a été représentée (oscillations absentes). Ce tracé montre une structure fine en dents de scie qui provient du renouvellement des gouttes.

Polarographie par échantillonnage.
Cette technique augmente la sensibilité de la polarographie à courant continu classique en atténuant le courant capacitif. La zone en grisé correspond à l’instant et à la durée de la mesure de courant.

Polarographie à impulsions.
Techniques NPP et DPP. Le dessin de l’extrémité de l’électrode de travail avec une goutte en formation montre à quel moment les mesures sont faites (voir les flèches a et b). Exemples de dosage : le dosage de l’acide ascorbique (vitamine C) dans un jus de fruit (voir conditions) correspond à une oxydation sur l’électrode de travail.

Polarographie à signaux carrés.
On superpose au niveau de chaque palier de potentiel E un signal carré périodique d’amplitude ΔEA = 2,5mV environ et de période égale à 40 ms. L’écart ΔE (marche entre chaque palier) est d’environ 12mV pour une durée de 0,5 à 10 s. L’intensité du courant est mesurée durant la zone grisée au niveau des points hauts (i+) et au niveau des points bas (i-) du signal périodique carré. La différence d’intensité i+/i- est portée en fonction du potentiel du palier. (d’après un document de la société Métrohm).

Détermination de cations à l’électrode à film de mercure par voltampérométrie inverse anodique.
Programmation linéaire en deux étapes du potentiel de l’électrode de travail dans l’analyse de métaux présents dans une eau de mer. L’électrode est constituée par un petit disque de carbone vitreux sur lequel un film de mercure (10 − 50 μm) est généré au moment de l’analyse en ajoutant à la solution échantillon 0,2 mL de nitrate mercurique (1 g/L).

Coulomètre pour déterminer la teneur en eau selon Karl Fischer.
Appareil C30S de la société Mettler Toledo.

Les électrodes d’un coulomètre de Karl Fischer.
Le diaphragme a pour but d’éviter que les ions réduits à la cathode (ou H2) ne réduisent éventuellement des composés oxygénés présents dans l’échantillon qui pourraient générer de l’eau.

Détection voltampérométriques en CLHP et ECHP.
a) Deux modèles de cellules voltampérométriques. L’électrode indicatrice, en graphite poreux, de grande surface, travaille dans les conditions coulométriques. La circulation de la phase mobile au niveau de l’électrode indicatrice assure le renouvellement des espèces électroactives ; b) détail au niveau de l’extrémité du capillaire en ECHP. L’électrode de travail reçoit les ions sortant du capillaire. La cellule n’est pas matérialisée. Elle se trouve réunie avec le compartiment cathodique de l’appareil. En dehors des phénols, amines aromatiques et thiols, peu de molécules analytiquement importantes sont électroactives.

Une cellule à deux électrodes concentriques, de type Clark pour le dosage du dioxygène.
La membrane en téflon, perméable au dioxygène, est très proche de la cathode pour que la double diffusion, à travers la membrane et ensuite dans le film liquide, conduise à un signal stable au bout de 10 à 15 secondes.

Détecteurs ampérométriques pour gas (Amperometric Gas Sensors).
Assortiment de capteurs de la société ATMI Sensoric Div. (Ger.). Modèle de détecteur GasAlertMax3-DL de la société BW Technologies (Can.) ; la photo centrale montre le compartiment où sont rangés 4 capteurs de gaz spécifiques.

Cellule à deux électrodes pour le dioxygène.
L’anode en plomb se transforme progressivement en oxyde de plomb. Les ions OH− migrent de l’électrode de travail vers l’électrode auxiliaire par le conducteur ionique tandis que les électrons suivent le chemin inverse par le circuit extérieur. Ce capteur est en quelque sorte une batterie plomb/air qui s’use avec le temps, comme toute batterie.

Cellules à 2 et à 3 électrodes pour le monoxyde de carbone.
La transformation électrochimique consomme du dioxygène qui doit être présent dans la cellule. Dans le montage avec électrode de référence, le potentiostat compense de manière électronique la valeur de l’intensité détectée, afin d’augmenter la plage de linéarité du capteur.

Oxydation du glucose par la glucose oxydase.
Le schéma réactionnel représenté correspond au bilan de cette réaction pour laquelle le dioxygène n’intervient que pour régénérer le cofacteur (même nombre d’atomes d’oxygène pour le glucose et la lactone). Détail de la partie active d’une membrane de reconnaissance chimique. Le film de polycarbonate fait barrière aux macromolécules éventuellement présentes dans l’échantillon et le film d’acétate de cellulose laisse passer les petites molécules H2O2 et O2.

Dosage ampérométrique du glucose.
Première génération basée sur la présence naturelle du dioxygène et exemple de seconde génération utilisant comme médiateur l’ion ferricyanure. Exemple de glucomètre pour déterminer le taux de glucose dans le sang. La bandelette qui intègre électrodes et membrane est à usage unique. Le volume de sang nécessaire, de l’ordre de 0,5 μL, est défini par la bandelette ellemême au moyen de l’électrode de remplissage (trigger electrode) qui délimite la cavité réactive (modèle Optium de la société Abbott).

Statistique faisant état de la répartition moyenne du temps passé pour faire une analyse par chromatographie.
La préparation des échantillons représente généralement une fraction importante du temps total consacré à l’analyse.

Extraction en phase solide.
Séparation d’un analyte de la matrice. Dans cet exemple l’analyte est le seul composé retenu. On distingue les étapes : a), b) activation et rinçage de l’adsorbant avant emploi ; c) dépôt d’un volume donné de l’échantillon ; d) élimination des interférences ; e) récupération de l’analyte par percolation. À droite, chromatogramme (CPG) obtenu après traitement par cette séquence d’1 mL de café. En bas, cartouches d’extraction en phase solide (SPE) ; on notera qu’elles ont une partie réservoir de 1 à 3 mL. À droite, préparateur d’échantillons (modèle Aspec XL4 reproduit avec l’autorisation de la société Gilson).

Principe de l’immuno-extraction.
Les différentes étapes du procédé classique sur un support greffé avec un anticorps adapté aux molécules « carrées ». Après l’étape de fixation (2), puis de rinçage (3), l’élution de l’analyte est faite avec un solvant organique (étape 4).

Procédés de micro extraction.
a) Micro extraction utilisant une fibre d’adsorption ; b) micro extraction avec une seule goutte de solvant organique.

Extraction d’un gaz.
Principe d’une colonne d’extraction gaz /solide – Réaction chimique mise en jeu pour dériver un aldéhyde (test d’atmosphère polluée, document Supelco Inc.).

Espace de tête en mode statique et en mode dynamique.
Dans le mode statique, l’échantillon est placé dans un flacon pressurisé. Quand l’équilibre thermodynamique est atteint, a lieu le prélèvement en vue de l’analyse par CPG. Dans le mode dynamique, un gaz vecteur entraîne, dans un premier temps, les composés volatils vers un piège (purge and trap), puis dans un second temps le piège est désorbé thermiquement pour libérer les composés qui y ont été concentrés en vue de l’analyse par CPG (modèle Versa, analyseur en mode statique et modèle HT3 mode statique et dynamique de la Société Teledyne-Tekmar).

Extraction par fluide supercritique.
Comparaison des forces d’élution du CO2 par rapport aux solvants usuels (échelle de Hildebrand) en fonction de la température et de la pression. L’extraction en phase supercritique est une méthode automatisable qui devient un investissement rentable dès lors que le nombre d’échantillons est important.

